Chain-end- and in-chain-functionalized AB diblock copolymers as key building blocks in the synthesis of well-defined architectural polymers
Akira
Hirao
*,
Kota
Murano
,
Toshiyuki
Oie
,
Masahiro
Uematsu
,
Raita
Goseki
and
Yuri
Matsuo
Polymeric and Organic Materials Department, Graduate School of Science and Engineering, Tokyo Institute of Technology, S1-6, 2-12-1, Ohokayama, Meguro-ku, Tokyo 152-8552, Japan
First published on 4th March 2011
Abstract
This paper reviews the precise synthesis of architectural polymers by methodologies utilizing either chain-end- or in-chain-functionalized AB diblock copolymers as efficient key building blocks. Architectural polymers herein synthesized are miktoarm star-branched polymers, exact graft copolymers, high-density comblike polymers, and alternative and sequential multiblock copolymers. Chain-end- and in-chain-functionalized AB diblock copolymers and, in some cases, core-functionalized ABC star-branched polymers utilized as building blocks are prepared by living anionic polymerization in conjunction with specially functionalized agents and linked in manners suitably designed for each architectural polymer to successfully synthesize such structurally complex polymers. The resulting polymers are all well-defined in structure and precisely controlled in chain length.
 Akira Hirao | Akira Hirao is currently a Professor of Polymeric and Organic Materials Department, Tokyo Institute of Technology, Tokyo, Japan. He completed his doctorate degree at Tokyo Institute of Technology in 1975 under the supervision of Professor Noboru Yamazaki. Following two years in post-doctoral work, he joined the Polymer Chemistry faculty of Tokyo Institute of Technology. Following Assistant and Associate Professors, he promoted to his present position in 1996. His research interests are focused on precise synthesis of functional (co)polymers and architectural polymers including block, graft, star-branched, and hyperbranched polymers by means of living anionic polymerization. |
 Kota Murano | Kota Murano was born in 1986 in Tokyo. He received his BS degree in polymer chemistry from Tokyo Institute of Technology under the supervision of Professor Akira Hirao in 2009. He is pursuing a masteral study focus on the precise synthesis of graft copolymers. |
 Toshiyuki Oie | Toshiyuki Oie was born in 1985 in Hukuoka. He received his BS degree in polymer chemistry from Tokyo Institute of Technology under the supervision of Professor Akira Hirao in 2009. He is currently pursuing his masteral study in the field of the synthesis of (AB)n multiblock copolymers. |
 Masahiro Uematsu | Masahiro Uematsu was born in Urawa, Japan, in 1986. He received his BSc degree in polymer chemistry from Tokyo Institute of Technology in 2010 under the supervision of Prof. Akira Hirao. He is currently pursuing his masteral studies in the field of precise synthesis of graft copolymers. |
 Raita Goseki | Raita Goseki received his M S in polymer chemistry from Tokyo Institute of Technology in 2005 under the supervision of Professor Masa-aki Kakimoto. He joined Hirao's laboratory as an Assistant Professor in 2010. His research interests are the precise synthesis and phase-selective chemistry in block copolymer system and the synthesis of star-branched polymers and block copolymers for applications in nano science and technology. |
 Yuri Matsuo | Yuri Matsuo was born in 1987 in Chiba, Japan. She is currently a bachelor student at Tokyo Institute of Technology, under the direction of Prof. Akira Hirao. Her interest is synthesis of ABA′ type asymmetrical triblock copolymers by means of living anionic polymerization. |
Introduction
Block copolymers, graft copolymers, and star-branched polymers are representative examples of “architectural polymers” and have been widely studied for a long time because of their synthetic challenge as well as their interesting properties and behavior in solution, melt, and solid states, different from corresponding linear polymers.1–3 In the case of polymers composed of chemically different segments, they are in general thermodynamically immiscible to phase-separate at the molecular level, followed by self-organizing, resulting in the formation of nanosize periodically ordered suprastructures and supramolecular assemblies. Such morphological nano-objects currently have many potential applications in the fields of nanoscience and nanotechnology.4–6Architectural polymers are generally synthesized by building up two or more polymer segments end-to-end or at the branching points via linking reactions. Each polymer segment used as a building block possesses a reaction site capable of linking with another polymer chain at an appropriate place(s), such as chain-end and in-chain positions. If AB diblock copolymers have a reaction site either at the chain-end or in-chain position, they can also act as new building blocks to synthesize architectural polymers in similar manners. With the use of such chain-end- and in-chain-functionalized AB diblock copolymers instead of single-polymer segments, the number of reaction steps required for the synthesis may be basically reduced by half, and new molecular design may make it possible to synthesize structurally complex architectural polymers which are difficult to synthesize by conventional procedures.
The choice of a living polymerization system is the other important factor to synthesize architectural polymers with well-defined structures. Although various living/controlled polymerization systems have been developed in the past three decades, the living anionic polymerization of styrene, 1,3-diene monomers, 2-vinylpyridine, alkyl methacrylates, and their derivatives with certain functional groups is still the best system due to the following features:7–9 (1) molecular weight can be precisely controlled in a wide range from 103 to even 106 g mol−1, (2) extremely narrow molecular weight distributions are attained. The Mw/Mn values are usually less than 1.05, and (3) their living anionic polymers have highly reactive chain-end anions which are capable of reacting with a variety of electrophiles in high yields, but stable enough under appropriate conditions. Such features are ideally suited for the synthesis of well-defined architectural polymers.
In this review article, we report on the precise synthesis of architectural polymers by the above living anionic polymerization-based methodologies, in which either chain-end- or in-chain-functionalized AB diblock copolymers are employed as key building blocks. The architectural polymers herein synthesized are miktoarm star-branched polymers, exact graft copolymers, high-density comblike polymers, and alternate and sequential multiblock polymers.
Miktoarm star-branched polymers
The first successful example of architectural polymer synthesis by utilizing chain-functionalized AB diblock copolymers as building blocks was the synthesis of a 3-arm ABC miktoarm star-branched polymer (Scheme 1).10 In this synthesis, an in-chain-silyl chloride-functionalized AB diblock copolymer was first prepared by a reaction of poly(isoprenyl)lithium (PILi) with a large excess of MeSiCl3, followed by evaporation of the unreacted excess MeSiCl3, and a subsequent reaction with polystyryllithium (PSLi) to link the two chains. Finally, poly(1,3-butadienyl)lithium (PBLi) was reacted with the resulting in-chain-silyl chloride-functionalized PI-b-PS to afford a 3-arm ABC miktoarm star-branched polymer composed of PI, PS, and PB segments (Mn = 45.4 kg mol−1 and Mw/Mn = 1.03). Almost at the same time, another strategy using an AB diblock copolymer having a 1,1-diphenylethylene (DPE)-derived anion between the A and B blocks, in-chain-(DPE anion)-functionalized AB diblock copolymer, was reported for the synthesis of another 3-arm ABC miktoarm star-branched polymer (Scheme 2).11 The (DPE anion)-functionalized building block was prepared by an addition reaction of PSLi to a chain-end-DPE-functionalized poly(dimethylsiloxane) (PDMS). The polymer anion thus prepared was used as a macroinitiator in the polymerization of tert-butyl methacrylate (tBMA), resulting in a 3-arm ABC miktoarm star composed of PS, PDMS, and PtBMA segments (Mn = 135 kg mol−1 and Mw/Mn = 1.05). Some other 3-arm ABC stars were also synthesized in similar manners.12–14 Thus, in-chain-functionalized AB diblock copolymers with a silyl chloride function and DPE-derived anion were acted as efficient building blocks to synthesize well-defined ABC and even 4-arm ABCD miktoarm star-branched polymers.15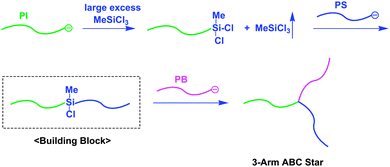 | ||
| Scheme 1 Synthesis of ABC star-branched polymer by the methodology utilizing in-chain-SiCl-functionalized AB diblock copolymer as the building block. | ||
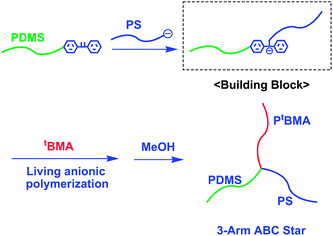 | ||
| Scheme 2 Synthesis of ABC star-branched polymer by the methodology utilizing in-chain-(DPE anion)-functionalized AB diblock copolymer as the building block. | ||
Hirao et al. have extended the above-mentioned methodologies to a general and versatile procedure, with which a variety of many-armed and many compositional miktoarm star-branched polymers have been synthesized.16–18Scheme 3 shows the first proposed methodology utilizing an in-chain-DPE-functionalized AB diblock copolymer as a building block.19,20 A living poly(A) was reacted with 1-(4-(3-bromopropyl)phenyl)-1-phenylethylene (1) to introduce a DPE function at the chain-end. Then, a living poly(B) was reacted with the DPE terminus of poly(A) to connect two chains with the generation of a DPE-derived anion at the linking point. The DPE function was re-introduced between the poly(A) and poly(B) segments by reacting 1 with the DPE-derived anion. Finally, a living poly(C) was reacted with the resulting in-chain-DPE-functionalized AB diblock copolymer to afford an ABC miktoarm star-branched polymer. Thus, the in-chain-DPE-functionalized AB diblock copolymer acted as an efficient building block in this synthesis, similar to the above-introduced in-chain-functionalized polymers.
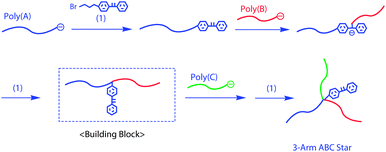 | ||
| Scheme 3 Synthesis of ABC star-branched polymer by the methodology utilizing in-chain-DPE-functionalized AB diblock copolymer as the building block. | ||
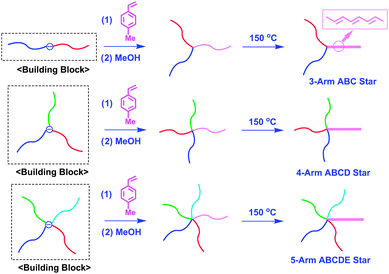
Since the resulting ABC star-branched polymer still possesses a DPE-derived anion at the core, the DPE function can be re-introduced at the core by treatment with 1. A further addition reaction of a living poly(D) to the resulting core-DPE-functionalized ABC star gave a 4-arm ABCD star-branched polymer. By repeating the same reaction sequence, a 5-arm ABCDE star could also be obtained. In such syntheses, both core-DPE-functionalized ABC and ABCD star-branched polymers acted as new building blocks (Scheme 4). Thus, the proposed methodology is more effective in star polymer synthesis than the previous methodologies mentioned above, since it enables the successive synthesis of a series of miktoarm stars of not only 3-arm ABC, but also 4-arm ABCD, 5-arm ABCDE, and so on to possibly more-armed star types. Typically, a new 5-arm ABCDE miktoarm star-branched polymer, composed of PS, poly(4-methoxystyrene) (PMOS), poly(4-methylstyrene) (PMS), poly(4-trimethylsilylstyrene) (PMSiMS), and poly(4-tert-butyldimethylsilyloxystyrene) (PBMSiOS), convertible to a poly(4-vinylphenol) segment,21 was successfully synthesized by this methodology (Mn = 55.5 kg mol−1 and Mw/Mn = 1.03).
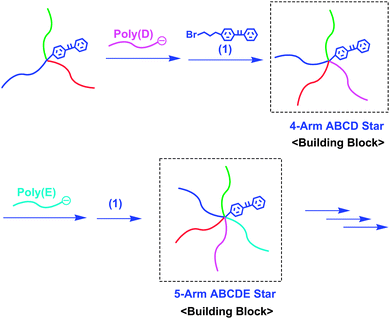 | ||
| Scheme 4 Synthesis of ABCD and ABCDE star-branched polymers by the methodology utilizing core-DPE-functionalized ABC and ABCD stars as building blocks. | ||
In the above synthesis, the intermediate AB block copolymer anion and star-branched polymers having DPE-derived anions at their cores can also be utilized as building blocks for the synthesis of interesting functional star-branched polymers. As shown in Scheme 5, a series of novel 3-arm ABC and 4-arm ABCD, followed by 5-arm ABCDE stars possessing π-conjugated conductive rod-like poly(acetylene) segments were successively synthesized by the living anionic polymerization of 4-methylphenyl vinyl sulfoxide (MPVS) with the above intermediate polymer anions, followed by thermal treatment at 150 °C.22,23 The resulting 5-arm ABCDE star composed of PS, PαMS, PMSiS, PMOS, and PMPVS (equivalent to poly(acetylene)) segments was observed to possess a predictable molecular weight and a narrow molecular weight distribution (Mn = 50.4 kg mol−1 and Mw/Mn = 1.03). Mavroudis and Hadjichristidis successfully synthesized a 4-arm ABCD star-branched polymer by a linking reaction of an in-chain-DPE-functionalized PI-b-PDMS with PSLi, followed by the polymerization of 2-vinylpyridine (2VP) (Mn = 75.9 kg mol−1 and Mw/Mn = 1.06) (Scheme 6).24 He et al. also synthesized a 4-arm ABCD star-branched polymer in a similar manner by the linking reaction of an in-chain-DPE-functionalized PS-b-PtBMA with PαMSLi and subsequent polymerization of 4-vinylpyridine.25
 | ||
| Scheme 5 Synthesis of ABC, ABCD and ABCDE star-branched polymers by the methodology utilizing (DPE anion)-functionalized polymers as building blocks. | ||
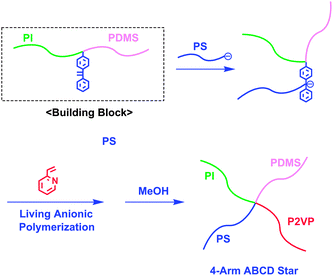 | ||
| Scheme 6 Synthesis of ABCD star-branched polymer by the methodology utilizing in-chain-DPE-functionalized AB diblock copolymer as the building block. | ||
An in-chain-(DPE anion)-functionalized PS-b-PI was quantitatively converted to the corresponding in-chain-NH2-functionalized PS-b-PI by treatment with 1-(3-bromopropyl)-2,2,5,5-tetramethyl-aza-2,5-disilacyclopentane, followed by deprotection of the silyl groups (Scheme 7).26 This NH2-functionalized block polymer could initiate the living anionic polymerization of NCA to result in miktoarm stars having rigid helical polypeptide segments, resulting in a 3-arm ABD star-branched polymer composed of PS, PI, and poly(γ-benzyl-L-glutamate) (Mn = 34.2 kg mol−1 and Mw/Mn = 1.09).27 Thus, the intermediate polymer anions and the NH2-modified block polymer served as macroinitiators (also regarded as building blocks) in the living anionic polymerization of appropriate monomers to synthesize two interesting functional miktoarm star-branched polymers.
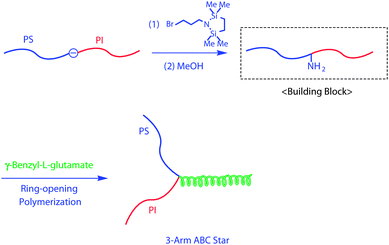 | ||
| Scheme 7 Synthesis of ABC star-branched polymer by the methodology utilizing in-chain-NH2-functionalized AB diblock copolymer as the building block. | ||
By combining in-chain-(DPE anion)-functionalized AB diblock copolymers with chain-end-(BnBr)n-functionalized polymers, a further star polymer synthesis is possible.28–30 For instance, an in-chain-(DPE anion)-functionalized AB diblock copolymer was coupled with a chain-end-benzyl bromide (BnBr)-functionalized poly(C) to afford an ABC miktoarm star (Scheme 8). Similarly, a 5-arm A2B2C star was synthesized by a reaction of the AB block copolymer anion with a chain-end-(BnBr)2-functionalized poly(C) (also see Scheme 8). Both chain-end-BnBr- and chain-end-(BnBr)2-functionalized poly(C)s were prepared by an addition reaction of 1-phenyl-1-(3-tert-butyldimethylsilyloxymethylphenyl)ethylene (2) and 1,1-bis(3-tert-butyldimethylsilyloxymethylphenyl)ethylene (3) to a living poly(C) and subsequent transformation of the introduced 3-tert-butyldimethylsilyloxymethylphenyl (SiOMP) group(s) to BnBr function(s) by treatment with Me3SiCl/LiBr. The addition reaction and subsequent transformation reaction were observed to proceed cleanly and quantitatively.
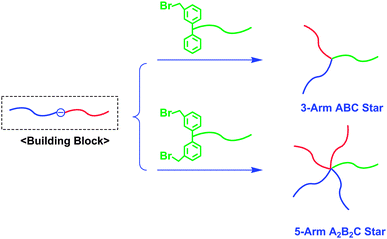 | ||
| Scheme 8 Synthesis of ABC and A2B2C star-branched polymers by the methodology utilizing in-chain-(DPE anion)-functionalized AB diblock copolymer as the building block. | ||
The number of BnBr termini was readily increased by a reaction of chain-end-(BnBr)2-functionalized poly(C) with a functionalized anion prepared from 3 and sec-BuLi, followed by a transformation reaction (Scheme 9).31 Surprisingly, the resulting chain-end-(BnBr)4-functionalized poly(C) was quantitatively coupled with a in-chain-(DPE anion)-functionalized AB diblock copolymer without difficulty to afford a 9-arm A4B4C star. More surprisingly, even an ABC star-branched polymer anion at the core could also be quantitatively reacted with the same chain-end-(BnBr)4-functionalized polymer. As a result, a new complex 13-arm A4B4C4D miktoarm star-branched polymer was obtained (see also Scheme 9).30 The resulting star-branched polymers are all well-defined in structure, as listed in Table 1. Thus, the methodology combining AB diblock copolymer anions (or even 3-arm ABC star polymer anions) with chain-end-(BnBr)n-functionalized polymers (n = 1, 2, or 4) provides an excellent procedure for the synthesis of complex miktoarm star-branched polymers, most of which are new and synthetically difficult by any other method.
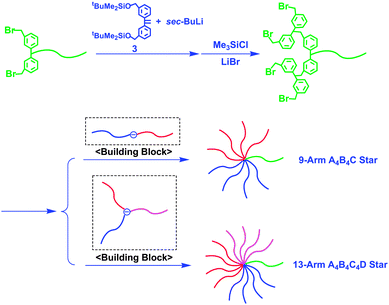 | ||
| Scheme 9 Synthesis of A4B4C and A4B4C4D star-branched polymers by the reaction of chain-end-(BnBr)4-functionalized polymers with in-chain-(DPE anion)-functionalized polymers as building blocks. | ||
| Typea | M n × 10−3/g mmol−1 | M w/Mn | |
|---|---|---|---|
| Calcd | SLS | SEC b | |
| a A, B, CA′, B′, C′, and D′ segments are PS, PI, PαMS, PSIS, PMOS, PMS, and PS respectively. b Determined by SEC equipped with triple detectors. | |||
| 3-ArmABC | 30.5 | 31.5 | 1.04 |
| 5-ArmA2B2C | 26.8 | 24.6 | 1.10 |
| 9-ArmA′4B′4C′ | 73.6 | 77.9 | 1.03 |
| 13-ArmA′4B′4C′4D′ | 134 | 133 | 1.02 |
Hirao et al. also proposed a similar effective methodology using in-chain-BnBr-functionalized AB diblock copolymers as building blocks (Scheme 10).32
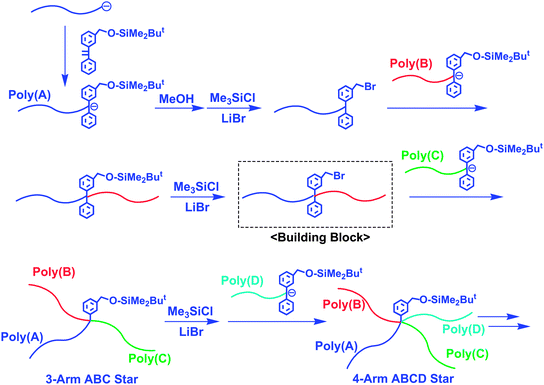 | ||
| Scheme 10 Synthesis of ABC and ABCD star-branched polymers by the methodology utilizing in-chain-BnBr-functionalized AB diblock copolymer as the building block. | ||
A key building block is prepared as follows: a living anionic poly(A) was end-capped with 2 to introduce a SiOMP group at the chain-end, which was subsequently transformed into a BnBr function by treatment with Me3SiCl/LiBr. Then, the resulting chain-end-BnBr-functionalized poly(A) was reacted with a living poly(B) end-capped with 2 to connect two polymer chains with the introduction of SiOMP group at the linking point. Treatment with Me3SiCl/LiBr gave an in-chain-BnBr-functionalized AB diblock copolymer. By linking the resulting in-chain-BnBr-functionalized AB block copolymer with a living poly(C) end-capped with 2, a 3-arm ABC miktoarm star-branched polymer composed of PS, PαMS, and PMS segments was quantitatively obtained (Mn = 35.0 kg mol−1 and Mw/Mn = 1.02).
This methodology also enables the successive synthesis of star-branched polymer to result in a 4-arm ABCD miktoarm star-branched polymer, since the ABC star-branched polymer still possesses the SiOMP group at the core. The resulting well-defined 4-arm ABCD star was composed of PS, PαMS, PMS, and PMMA segments (Mn = 47.9 kg mol−1 and Mw/Mn = 1.02). Thus, the SiOMP group is designed in such a way that it is stable toward living anionic polymers and quantitatively transformed into a BnBr function usable as the next reaction site.32,33
In order to successively synthesize a series of miktoarm star-branched polymers by the above methodology using in-chain- or (core-) DPE- and -BnBr-functionalized polymers, the living anionic polymer used in each reaction step is always required to react either with the DPE function in the polymer chain or with 2 of the DPE derivative (see Schemes 4 and 10). Therefore, usable living polymers are limited only to highly reactive living anionic polymers of styrene, 1,3-butadiene, isoprene, and their derivatives. Typically, PSLi, PILi, PBLi, PMSLi, PMOSLi, PαMSLI, and PMSiSLi are used. Furthermore, living anionic polymers of 4-(tert-butyldimethylsilyloxy)styrene and 4-(bis(N,N-trimethylsilyl)amino)styrene, equivalent to poly(4-vinylphenol) and poly(4-vinylaniline) segments,21,34 are also usable. With this methodology, not only ABCD, ABCDE, etc. stars, but also A3B, A2BC, or any other star types can be successively synthesized by appropriately changing the living anionic polymer in the linking reaction step.
On the other hand, less reactive living anionic polymers of 2VP, MMA, tBMA, and ethylene oxide (EO) can also be used in the linking reaction step with either in-chain-BnBr-functionalized AB diblock copolymers or core-BnBr-functionalized stars to afford miktoarm stars. These living polymers were quantitatively reacted with BnBr reaction sites to introduce one more polymer chain. In the case of in-chain-DPE-functionalized diblock copolymers and core-DPE-functionalized stars, treatment of such DPE-functionalized polymers with sec-BuLi to convert them to the corresponding DPE anion-functionalized polymers, followed by the living anionic polymerization of the above monomers, gave star-branched polymers having one more additional arm.11–14,22–25 It should be mentioned, however, that the successive synthesis could not continue in either case because the next reaction sites were not introduced.
Recently, a new in-chain-(α-phenyl acrylate)-functionalized AB diblock copolymer composed of PS (A) and poly(ferrocenylmethyl methacrylate) (PFeMMA) (B) segments was prepared by the sequential block copolymerization of styrene, 2, and FeMMA, followed by treatment first with (C4H9)4NF and then with α-phenyl acrylate under Mitsunobu reaction conditions.35 By this treatment, the SiOP group was converted to an α-phenyl acrylate function. This polymer can be used as a building block for star polymer synthesis. For example, two interesting functional 3-arm ABC miktoarm star-branched polymers composed of PS, PFeMMA, and either PMMA or P2VP segments were synthesized by the reaction of the in-chain-(α-phenyl acrylate)-functionalized PS-b-PFeMMA with each of the corresponding living polymers of MMA and 2VP (Scheme 11). Moreover, two α-phenyl acrylate functions were introduced between the PS and PMMA blocks by using 3 instead of 2. A new 4-arm ABC2 star-branched polymer composed of PS, PMMA, and PFeMMA segments was quantitatively obtained by linking it with the living polymer of FeMMA. The resulting star-branched polymers are all well-defined in structure and precisely controlled in chain length, as listed in Table 2.
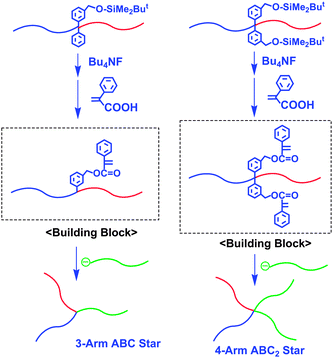 | ||
| Scheme 11 Synthesis of ABC and ABC2 star-branched polymers by the methodology utilizing in-chain-(α-phenyl acrylate)n-functionalized AB diblock copolymers (n= 1 and 2) as building blocks. | ||
Hadjichristidis et al. have recently demonstrated the successful synthesis of 4-arm A2B2, A2BC, and 5-arm A2B2C miktoarm stars by the combination of chlorosilane and benzyl chloride (BnCl) linking chemistry.36 The A, B, and C arms were PDMS, PS, and PI segments. The synthetic outline is given in Scheme 12, where an in-chain-BnCl-functionalized PDMS is used as the building block. It was prepared by a selective linking reaction of the lithium silanolate terminus of PDMS with the two Si–Cl reaction sites of dichloro(2-(chloromethylphenyl)ethyl)methylsilane, leaving the BnCl function intact. The A2B2 and A2BC miktoarm star-branched polymers (Mw = 88.7 and 96.4 kg mol−1 and Mw/Mn = 1.05 and 1.02) were synthesized by a reaction of the in-chain-BnCl-functionalized PDMS with the in-chain-(DPE-anion)-functionalized PS and PS-b-PI. As often mentioned, a core-(DPE anion)-functionalized 3-arm A2B star composed of PS (A) and PI (B) segments is prepared by an addition reaction of PSLi to the in-chain-DPE-functionalized PS-b-PI. The linking reaction of this star polymer anion with the in-chain-BnCl-functionalized PDMS gave a 5-arm A2B2C star (Mw = 109 kg mol−1 and Mw/Mn = 1.02) (see also Scheme 12).
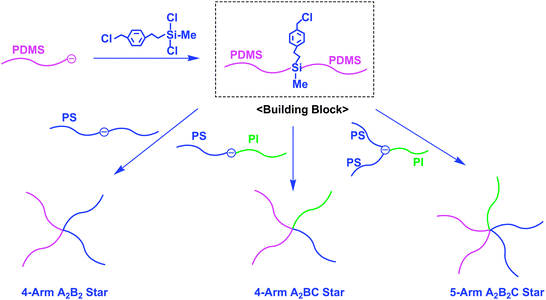 | ||
| Scheme 12 Synthesis of A2B2, A2BC, and A2B2C star-branched polymers by the methodology utilizing in-chain-BnCl-functionalized PDMS as the building block. | ||
In summary, a variety of miktoarm star-branched polymers were successfully synthesized by a methodology utilizing in-chain-functionalized AB diblock copolymers and, in some cases, core-functionalized star-branched polymers as building blocks. Most of the miktoarm star-branched polymers synthesized by this methodology are new structurally complex stars which are synthetically difficult by any other methods. The resulting miktoarm star-branched polymers were all well-defined in structure and precisely controlled in chain length. Their molecular weight distributions were always narrow (Mw/Mn ≤ 1.05). Thus, the methodology utilizing in-chain-functionalized AB diblock copolymers provides a general and versatile procedure for the synthesis of a variety of well-defined miktoarm star-branched polymers.
Recently, the synthesis of miktoarm star-branched polymers by similar methodologies, combining living/controlled radical polymerization with click reactions was developed by several research groups. Although the procedures are simpler and more convenient, the resulting polymers are not well-defined in structure at the present time, in comparison with those synthesized in this section.
Graft copolymers
A. Exact graft copolymers
The structure of the graft copolymer is defined by the following four variables: (1) molecular weight of main chain, (2) molecular weight of branch chain, (3) distance between branch chains, and (4) number of branch chains along with the main chain. An ideal graft copolymer, in which all of the four variables are perfectly controlled, has been named “an exact graft copolymer” by Paraskeva and Hadjichristidis.37 Although several attempts have been made to synthesize such ideal graft copolymers, most of those so far synthesized have not been completely controlled with respect to the above four variables.38–41Paraskeva and Hadjichristidis reported the first successful synthesis of an exact graft copolymer composed of a PI main chain and two PS branches by a stepwise iterative methodology using an in-chain-(DPE anion)-functionalized AB diblock copolymer as the building block.37 The synthetic outline is given in Scheme 13. The building block was prepared by the addition of PILi to 1,4-bis(phenylethenyl)benzene (4) to introduce the DPE function at the chain-end and a subsequent addition reaction of a stoichiometric amount of PSLi to the resulting chain-end-DPE-functionalized PI. The resulting diblock copolymer anion was used as a macroinitiator in the living anionic polymerization of isoprene. By repeating the same reaction sequence, an exact graft copolymer having two PS branch chains (Mn = 233 kg mol−1 and Mw/Mn = 1.08) was synthesized. Moreover, the same group has recently synthesized other exact graft PB having three PB branches by using a 3-arm A3 star-branched polymer anion as a building block (Scheme 14) (Mn = 187 kg mol−1 and Mw/Mn = 1.09).42 Recently, Hirao et al. developed an improved methodology to successfully synthesize a series of exact graft (PS)s having up to five PS branches (Mn = 124 kg mol−1 and Mw/Mn = 1.03).43
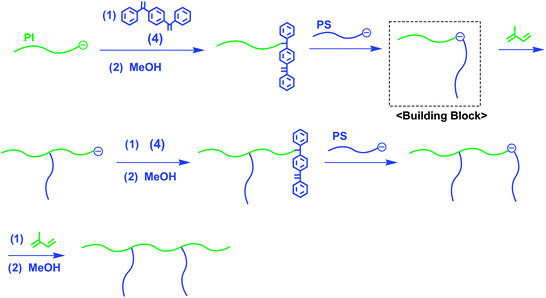 | ||
| Scheme 13 Synthesis of exact graft copolymer by the methodology utilizing in-chain-(DPE anion)-functionalized AB diblock copolymer as the building block. | ||
 | ||
| Scheme 14 Synthesis of exact graft poly(1,3-butadiene) by the methodology utilizing chain-end-(DPE anion)-functionalized A3 star as the building block. | ||
More recently, Hirao et al. have proposed a novel stepwise iterative methodology for the synthesis of a series of exact graft copolymers composed of a PMMA main chain and up to five PS branches.44 The methodology is illustrated in Scheme 15, where an in-chain-SiOMP-functionalized living AB diblock copolymer composed of PS and PMMA segments is employed as the key building block. The following three reaction steps are used in an iterative synthetic sequence: (1) anionic block copolymerization to prepare an in-chain-SiOMP-functionalized living PS-b-PMMA, (2) a transformation reaction of the introduced SiOMP group to the BnBr function, and (3) a linking reaction of the in-chain-SiOMP-functionalized living PS-b-PMMA with in-chain-BnBr-functionalized PS-b-PMMA. By repeating the above synthetic sequence five times, a series of exact graft copolymers with up to five PS branches were successfully synthesized. As listed in Table 3, the resulting graft copolymers are all well-defined in structure and precisely controlled in molecular weight.
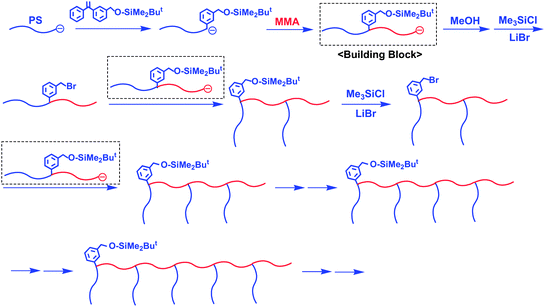 | ||
| Scheme 15 Synthesis of exact graft copolymers by the methodology utilizing in-chain-SiOMP-functionalized living AB diblock copolymer as the building block. | ||
In these polymers, the molecular weights of the main chain and each branch chain correspond to the total molecular weight of the PMMA blocks and the molecular weight of each PS block, respectively. The distance between branch chains is exactly equal to the molecular weight of each PMMA block. The number of branch chains can be determined by the number of synthetic sequences. Thus, the series of resulting polymers are target exact graft copolymers having two, three, four, and five branches. Since the final polymer possesses an SiOMP terminus, it may be possible to continue the iterative synthetic sequence to afford a series of exact graft copolymers with six and more branches. Thus, the methodology using in-chain-SiOMP-functionalized living AB diblock copolymers as key building blocks offers the potential of providing a general procedure.
B. High-density comblike copolymers
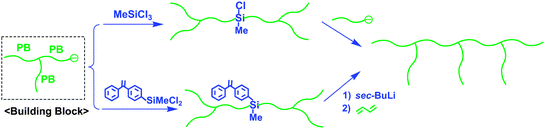
The poly(macromonomer), accessible by the living polymerization of macromonomers, is one of the ultimate graft copolymers carrying one branch per each repeating unit.45–48 Structurally similar graft copolymers were synthesized by a quantitative linking reaction of PSLi with either poly(2-chloroethyl vinyl ether)49 (prepared by living cationic polymerization) or poly(3-bromomethylstyrene)50 (prepared by living anionic polymerization of 3-tert-butyldimethylsilyloxymethylstyrene, followed by transformation with Me3SiCl/LiBr), which was based on the “grafting-onto” method.On the basis of successful synthesis by the grafting-onto method, Hirao et al. attempted to synthesize an extremely high-density graft copolymer having two different branch chains per each repeating unit by a linking reaction of poly(3-bromomethylstyrene) with an in-chain-(DPE anion)-functionalized AB diblock copolymer, often used as the building block in the star polymer synthesis mentioned above. However, the linking efficiency always remained at 50% under various conditions, possibly due to steric hindrance.
As an alternative main chain, poly(4-(3-(4-bromomethylphenyl)propyl)styrene) (poly(5)) was newly prepared by the living anionic polymerization of 4-(3-(4-tert-butyldimethylsilyloxymethylphenyl)propyl)styrene, followed by transformation of the SiOMP groups into BnBr functions (Scheme 16).51 Poly(5) is designed in such a way that the BnBr reaction site is separated by a 3-propylphenyl group from the PS main chain in order to reduce the steric hindrance. As expected, poly(5) (Mw = 6. 08 kg mol−1 and Mw/Mn = 1.02) was quantitatively linked with an in-chain-(DPE anion)-functionalized PS composed of two PS segments (Mw = 11.2 kg mol−1 (5.34/5.70)) in THF at −40 °C within 1 h to afford the target high-density comblike PS (Mw = 216 kg mol−1 and Mw/Mn = 1.02) carrying two PS branches per repeating unit. Moreover, a linking reaction of relatively high-molecular-weight poly(5) (Mw = 17.0 kg mol−1 and Mw/Mn = 1.02) with any of the in-chain-(DPE anion)-functionalized PSs with different molecular weights (Mw = 10.2 kg mol−1 (4.87/5.35), 27.5 kg mol−1 (21.8/5.70), and even 54.6 kg mol−1 (49.2/5.35)) proceeded without difficulty. Similarly, high-density comblike copolymers having PS and PI segments in each unit could be synthesized by the same linking reaction of poly(5) (Mw = 6.08 kg mol−1 and Mw/Mn = 1.02) with in-chain AB diblock copolymer (PS-b-PI) anions (Mw = 11.9 kg mol−1 (PS/PI = 6.47/5.40) and Mw = 26.0 kg mol−1 (PS/PI = 20.6/5.40)). All of the results are summarized in Table 4.
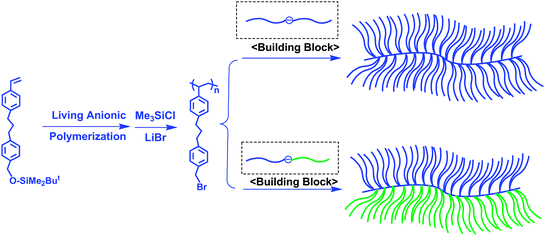 | ||
| Scheme 16 Synthesis of high-density comblike polymers by the methodology utilizing chain-end-(DPE anion)-functionalized A polymer and AB diblock copolymer as building blocks. | ||
| Typea | Main chain | Side chain | Branched polymer(Mn × 10−3/g mol−1) | |
|---|---|---|---|---|
| M n × 10−3/g mol−1 | M n × 10−3/g mol−1 | Calcd | Obsd | |
| a Combination of side chain polymers. | ||||
| PS/PS | 6.08 | 11.2(5.34/5.70) | 217 | 216 |
| PS/PS | 17.0 | 10.2(4.87/5.35) | 551 | 546 |
| PS/PS | 17.0 | 27.5(21.8/5.70) | 1480 | 1540 |
| PS/PS | 17.0 | 54.6(49.2/5.35) | 2942 | 3060 |
| PS/PI | 6.08 | 11.9(6.47/5.40) | 229 | 239 |
| PS/PI | 6.08 | 26.0(20.6/5.40) | 502 | 528 |
Thus, a linking reaction of poly(5) with either in-chain-(DPE anion)-functionalized PS or PS-b-PI provides a promising procedure for the synthesis of extremely high-density comblike copolymers carrying two branches in each repeating unit. It should be mentioned that the resulting densely branched polymers herein synthesized all appear to be star-like rather than rod-like in structure, estimating from the observed g' values which were obtained from the ratios of [η]branched polymer/[η]linear polymer.
Block polymers
A. Alternate multiblock copolymers
Block copolymers with well-defined structures are generally synthesized by the living anionic polymerization where two or more monomers are sequentially added to an appropriate anionic initiator. Among the monomers with similar reactivities (or electrophilicities), a growing chain-end anion formed from the first monomer initiates the polymerization of the second added monomer and vice versa. For example, the sequential addition of styrene and isoprene to sec-BuLi gives a well-defined AB diblock copolymer, PS-b-PI. A BA diblock copolymer, PI-b-PS, is also synthesized by the addition of two monomers in a reversed order. Thus, crossover generally occurs to such monomers and, therefore, not only AB and BA diblocks, but also ABA, ABAB, and even (AB)n multiblock copolymers can be synthesized by the sequential addition of A, B, A, B, and so on. In fact, several multiblock copolymers of styrene and either isoprene or α-methylstyrene were synthesized by several research groups52–55In contrast, the order of monomer addition is very critical among monomers with different reactivities in sequential living anionic polymerization. In general, a more reactive growing chain-end anion is produced by a less reactive monomer and vice versa because the electron-withdrawing effect of the substituent simultaneously influences both reactivities of the monomer and the growing chain-end anion. Accordingly, less reactive monomers should first be polymerized, followed by the polymerization of more reactive monomers. In the block copolymer of styrene and MMA, for instance, it is necessary first to polymerize styrene and then MMA is polymerized to prepare a block copolymer, PS-b-PMMA. The reverse addition of the two monomers affords only a PMMA homopolymer, because the growing chain-end enolate anion derived from MMA cannot initiate the polymerization of styrene. Thus, the synthesis of a multiblock copolymer of styrene and MMA is not possible by means of sequential polymerization.
This synthetic problem has been recently overcome by developing a new methodology which involves linking and chain-modification reactions in an iterative synthetic sequence.56 The outline of the methodology is illustrated in Scheme 17. In this synthesis, an α-terminal-(3-tert-butyldimethylsilyloxypropyl) (SiOP)-functionalized living anionic AB diblock copolymer composed of PS and PMMA blocks is employed as the building block. The α-functionalized living block copolymer was prepared by the sequential living anionic block copolymerization of styrene, DPE (used only for end-capping of the poly(styryl)anion to avoid ester attack), and MMA with 3-tert-butyldimethylsilyloxy-1-propyllithium (SiOPLi). Then, the α-SiOP terminus was deprotected, followed by esterification with α-phenyl acrylic acid, to convert it into an α-phenyl acrylate function. The resulting α-terminal-(α-phenyl acrylate)-functionalized AB diblock copolymer was reacted with an α-SiOP-functionalized living AB diblock copolymer prepared in advance, resulting in an α-SiOP-functionalized ABAB tetrablock copolymer. Since the two reaction steps were observed to quantitatively proceed, the same reaction sequence could be repeated three more times to successfully synthesize new (AB)3 hexablock, (AB)4 octablock, and (AB)5 decablock copolymers. The resulting alternate multiblock copolymers were all observed to possess predictable molecular weights, compositions, and narrow molecular weight distributions (see Table 5). It may be possible to continue the same synthetic sequence, since the (AB)5 decablock copolymer still possesses the α-SiOP terminus.
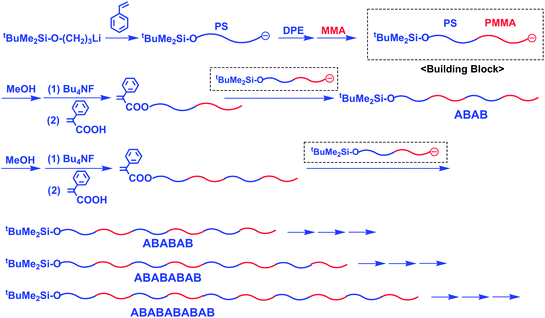 | ||
| Scheme 17 Synthesis of a series of alternate multiblock copolymers by the methodology utilizing α-chain-end-SiOP-functionalized living AB diblock copolymer as the building block. | ||
| Code | Type | M n × 10−3/g mol−1 | M w/Mn | Composition (wt%) | ||
|---|---|---|---|---|---|---|
| Calcd | RALLS | SEC a | Calcd | 1H NMR | ||
| a Determined by SEC equipped with triple detectors. | ||||||
| AB | Diblock | 10.5 | 11.4 | 1.03 | 50/50 | 48/52 |
| (AB)2 | Tetrablock | 26.4 | 28.2 | 1.03 | 48/52 | 47/53 |
| (AB)3 | Hexablock | 37.0 | 40.5 | 1.03 | 45/55 | 43/57 |
| (AB)4 | Octablock | 51.0 | 53.6 | 1.04 | 45/55 | 43/57 |
| (AB)5 | Decablock | 64.5 | 66.4 | 1.06 | 45/55 | 45/55 |
This iterative methodology also enables the synthesis of alternate (AB)n multiblock copolymers composed of a PS (A) segment and either PtBMA or P2VP as a B segment. For such a synthesis, both α-SiOP-functionalized PS-b-PtBMA and PS-b-P2VP living anionic block copolymers were employed as the building blocks. They worked satisfactorily to result in two series of alternate (AB)n multiblock copolymers composed of PS and either PtBMA or P2VP (n = 1, 2, and 3). The resulting polymers are well-defined in structure, as listed in Table 6.
| Polymer | Type | M n × 10−3/g mol−1 | M w/Mn | Composition (wt%) | |||
|---|---|---|---|---|---|---|---|
| A | B | Calcd | RALLS | SEC a | Calcd | 1H NMR | |
| a Determined by SEC equipped with triple detectors. | |||||||
| PS | P2VP | AB | 10.4 | 13.1 | 1.03 | 47/53 | 46/54 |
| PS | P2VP | ABAB | 24.4 | 25.8 | 1.03 | 47/53 | 46/54 |
| PS | P2VP | ABABAB | 41.5 | 46.6 | 1.06 | 48/52 | 46/54 |
| PS | PtBMA | AB | 11.3 | 12.7 | 1.04 | 52/48 | 50/50 |
| PS | PtBMA | ABAB | 24.3 | 25.3 | 1.04 | 49/51 | 49/51 |
| PS | PtBMA | ABABAB | 37.2 | 38.3 | 1.03 | 49/51 | 48/52 |
B. Sequential multiblock polymers
Since the first and second (or higher) block segments are thermodynamically incompatible in most block copolymers, an interesting morphology, based on molecular-level phase separation between the two blocks, followed by self-organization, is observed. The morphology of AB diblock copolymers has been widely studied and well established as the Molar's rule. Recently, several ABC triblock terpolymers have exhibited more interesting morphological features, moving the borders of the classical morphology map, because the addition of a third block tremendously increases the variety of new morphological suprastructures.57–60 Moreover, new interesting supramolecular assemblies have been created by ABC triblock terpolymers.61,62 Accordingly, it is expected that ABC triblock terpolymers and other sequential multiblock polymers will become promising heterophase materials next to AB diblock copolymers and that their basic morphological behavior will be studied in order to apply them in areas of nanoscience and nanotechnology in the near future.Among the monomers with similar reactivities, in addition to ABC block terpolymers, either ACB or BAC triblock terpolymers can be synthesized without problem. In practice, PS-b-PI-b-PB, PS-b-PB-b-PI, and PI-b-PS-b-PB were readily synthesized by the sequential addition of the corresponding monomers to sec-BuLi. As one of the most significant examples, an ABCD tetrablock quarterpolymer, PS-b-PB-b-poly(1,3-cyclopentadiene)-b-PI, was synthesized by Hadjichristidis et al. (Mn = 197 kg mol−1 and Mw/Mn = 1.08).63 A sequentially different tetrablock quarterpolymer, PS-b-PB-b-PI-b-poly(1,3-cyclopentadiene), could also be synthesized only by changing the addition order. Among the methacrylate monomers, a series of ABC, ACB, and BAC triblock terpolymers was obtained by living anionic block polymerization when tBMA, 2-(trimethylsilyloxy)ethyl methacrylate (SiHEMA), and 2-(perfluorobutyl)ethyl methacrylate were sequentially polymerized in this or a different order.64 In each of the resulting polymers, the trimethylsilyl protective group of the PSiHEMA block was removed by acid treatment to convert it to poly(2-hydroxyethyl methacrylate). 1H NMR and light scattering measurements of the resulting triblock terpolymers in selective solvents clearly indicate the formation of micelles. Interestingly, their aggregation numbers were strongly affected by the sequential order of the three segments.65
As mentioned before, among the monomers with different reactivities, the monomer addition order is very critical and limited in the synthesis of block copolymers because the reactivity of the growing chain-end anion is not always sufficient to initiate the polymerization of another monomer. For the possible molecular design and successful synthesis of block copolymers, the monomer reactivity estimated from the “e” value, as well as the pKa value of the conjugated acid of the growing chain-end anion, is a valuable guide. With these reactivities of growing chain-end anions and monomers in mind, several well-defined ABC triblock terpolymers, such as PS-b-P(2VP)-b-PtBMA (Mn = 136 kg mol−1 and Mw/Mn = 1.03), PI-b-P(2VP)-b-PEO (Mn = 91.0 kg mol−1 and Mw/Mn = 1.04), and PS-b-PI-b-PDMS (Mn = 70.2 kg mol−1 and Mw/Mn = 1.02), were successfully synthesized.66–68 Moreover, the synthesis of ABCD tetrablock quarterpolymers, PS-b-PI-b-P(2VP)-b-PtBMA and PS-b-PI-b-P(2VP)-b-PEO (Mn = 126 kg mol−1 and Mw/Mn = 1.06), and even a particular ABCDE pentablock quintopolymer, PS-b-PI-b-P(2VP)-b-PtBMA-b-PEO (Mn = 92.0 g mol−1 and Mw/Mn = 1.04), could also be achieved.69
On the other hand, block polymers sequentially different from the above block copolymers cannot be synthesized because of the above reason. In order to overcome such a problem, Hadjichristidis et al. have developed a quite effective strategy using chain-end-BnCl-functionalized ABC triblock terpolymers as the building blocks, where A, B, C are PS, PI, and PDMS, respectively (Scheme 18).70 The linking agent, 2-(chloromethylphenyl)ethyldimethyl chlorosilane (6), was designed to selectively react with the terminal silanolate anion of the triblock terpolymer to introduce the BnCl function at the chain-end. A living P2VP was linked with the PS-b-PI-b-PDMS to afford a new tetrablock quarterpolymer, PS-b-PI-b-PDMS-b-P2VP. Furthermore, a new pentablock quintopolymer, PS-b-PI-b-PDMS-b-PtBMA-b-P2VP, could also be synthesized by linking a living AB diblock copolymer, P2VP-b-PtBMA, with the same chain-end-BnCl-functionalized PS-b-PI-b-PDMS. The resulting block polymers all possessed predictable molecular weights (Mn = 100–160 kg mol−1) and narrow molecular weight distributions (Mw/Mn ≤ 1.05).71,72
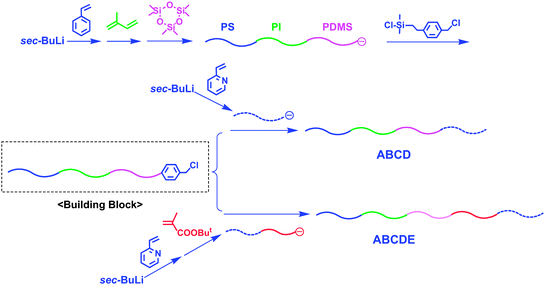 | ||
| Scheme 18 Synthesis of sequential multiblock polymers by the methodology utilizing chain-end-BnCl-functionalized ABC triblock terpolymer as the building block. | ||
The methodology effective for the synthesis of alternate multiblock copolymers developed in the preceding section is also applicable for the synthesis of sequentially different triblock terpolymers. Scheme 19 shows a typical synthetic outline of a triblock terpolymer, P2VP-b-PS-b-PMMA, synthetically difficult by sequential polymerization.73 For this synthesis, an α-terminal-(α-phenyl acrylate)-functionalized PS-b-PMMA also acts as the key building block. A living P2VP was then reacted with the α-terminal-(α-phenyl acrylate)-functionalized PS-b-PMMA to result in a quantitative formation of the target triblock terpolymer, P2VP-b-PS-b-PMMA. As expected, the polymer possessed a predictable molecular weight and a narrow molecular weight distribution (Mn = 39.7 kg mol−1 and Mw/Mn = 1.05). A new triblock terpolymer having a different sequence, PMMA-b-PS-b-P2VP, was also successfully synthesized in 100% yield by a linking reaction of living PMMA with the α-terminal-(α-phenyl acrylate)-functionalized PS-b-P2VP under the same conditions (Mn = 33.6 kg mol−1 and Mw/Mn = 1.04).
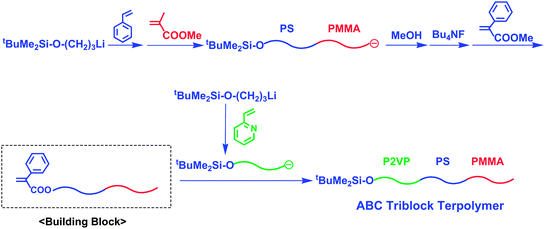 | ||
| Scheme 19 Synthesis of ABC triblock terpolymer by the methodology utilizing chain-end-(α-phenyl acrylate)-functionalized AB diblock copolymer as the building block. | ||
With this methodology, the synthesis of an ABA′ triblock copolymer, whose A blocks were different in molecular weight, was also possible (Scheme 20). For this synthesis, living PMMA (Mn = 20.0 kg mol−1) was prepared and in situ reacted with the α-terminal-(α-phenyl acrylate)-functionalized PS-b-PMMA (Mn = 21.6 kg mol−1 (Mn(PS)/Mn(PMMA) = 10.4 kg mol−1/11.2 kg mol−1)), resulting in a triblock ABA′ type copolymer, PMMA-b-PS-b-PMMA (Mn = 41.6 kg mol−1, Mw/Mn = 1.03). The PMMA segments on both sides were 20.0 kg mol−1 and 11.2 kg mol−1 in Mn value, respectively. Various tetra- or higher multiblock polymers can also be synthesized in a stepwise manner by repeating the synthetic sequence involving the chain-end modification to α-phenyl acrylate and a subsequent linking reaction, as illustrated in Scheme 21. Thus, the methodology combining a linking reaction of living anionic polymers with chain-end-functionalized block polymers as the building blocks provides a promising and excellent procedure, with which one can synthesize both alternate and sequential multiblock polymers possessing any sequential block order.
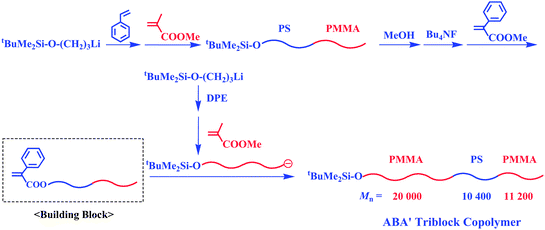 | ||
| Scheme 20 Synthesis of ABA′ triblock copolymer by the methodology utilizing chain-end-(α-phenyl acrylate)-functionalized AB diblock copolymer as the building block. | ||
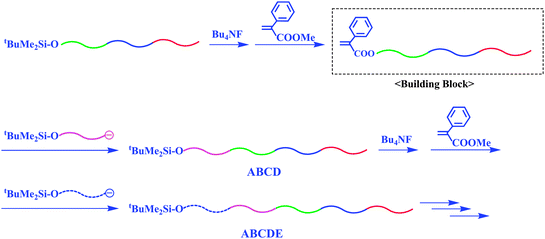 | ||
| Scheme 21 Synthesis of ABCD, ABCDE, and higher block polymers by the methodology utilizing chain-end-(α-phenyl acrylate)-functionalized AB diblock copolymer as the building block. | ||
Conclusions
This review focuses on the precise synthesis of structurally complex and synthetically difficult architectural polymers, including miktoarm star-branched polymers, exact graft copolymers, high-density comblike polymers, alternate and sequential multiblock polymers, by a methodology utilizing chain-end- or in-chain-functionalized AB block copolymers as the building blocks. Such architectural polymers are synthesized by procedures designed to link living anionic polymers with functional groups introduced at the chain-end and in-chain of block copolymers. With the use of chain-end- and in-chain-functionalized block copolymers, the number of reaction steps required for synthesis is reduced by half and a new molecular design makes it possible to synthesize several structurally complex architectural polymers which are synthetically difficult by conventional procedures.In most of the procedures herein introduced, the synthetic sequence may possibly be repeated in a stepwise manner by re-introducing the functional group in the final synthetic stage and thereby a series of more complex architectural polymers are synthesized. In such cases, the final product automatically corresponds to the building block of the next product.
Notes and references
- R. P. Quirk, in Anionic Polymerization: Principles and Application, ed. H. L. Hsieh and R. P. Quirk, Marcel Dekker, New York, 1996 Search PubMed.
- S. P. Meneghetti, P. J. Lutz and D. Rein, in Star and Hyperbranched Polymers, ed. M. K. Mishra and S. Kobayashi, Marcel Dekker, New York, 1999 Search PubMed.
- K. Matyjaszewski, in Macromolecular Engineering: Precise Synthesis, Materials Properties, Applications—Volume 1. Synthetic Techniques, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley-VCH, Weinheim, 2007 Search PubMed.
- C. Parka, J. Yoonb and E. L. Thomas, Polymer, 2003, 44, 6725 CrossRef CAS.
- M. A. Hillmyer, Adv. Polym. Sci., 2005, 190, 137 CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS.
- N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 857 CrossRef CAS.
- A. Hirao, S. Lokulnant and T. Ishizone, Prog. Polym. Sci., 2002, 27, 1399 CrossRef CAS.
- K. Sugiyama, K. Watanabe, A. Hirao and M. Hayashi, Macromolecules, 2008, 41, 4235 CrossRef CAS.
- H. Iatrou and N. Hadjichristidis, Macromolecules, 1992, 25, 4649 CrossRef CAS.
- T. Fujimoto, H. Zhang, T. Kazama, Y. Isono, H. Hasegawa and T. Hashimoto, Polymer, 1992, 33, 2208 CrossRef CAS.
- H. Hückstädt, A. Gopfert and V. Abetz, Macromol. Chem. Phys., 2000, 201, 296 CrossRef CAS.
- M. Nasser-Eddine, S. Reutenauer, C. Delaite, G. Hurtrez and P. Dumas, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1745 CrossRef CAS.
- A. Takano, S. Wada, S. Sato, T. Araki, K. Hirahara, T. Kazama, S. Kawahara, Y. Isono, A. Ohno, N. Tanaka and Y. Matsushita, Macromolecules, 2004, 37, 9941 CrossRef CAS.
- H. Iatrou and N. Hadjichristidis, Macromolecules, 1993, 26, 2479 CrossRef CAS.
- A. Hirao, M. Hayashi, Y. Tokuda, N. Haraguchi, T. Higashihara and S.-W. Ryu, Polym. J., 2002, 34, 633 CrossRef CAS.
- A. Hirao, K. Inoue, T. Higashihara and M. Hayashi, Polym. J., 2008, 40, 923 CrossRef CAS.
- T. Higashihara, K. Sugiyama, H.-S. Yoo, M. Hayashi and A. Hirao, Macromol. Rapid Commun., 2010, 31, 1031 CrossRef CAS.
- A. Hirao, M. Hayashi and T. Higashihara, Macromol. Chem. Phys., 2001, 202, 3165 CrossRef CAS.
- A. Hirao and T. Higashihara, Macromolecules, 2002, 35, 7238 CrossRef CAS.
- T. Higashihara, K. Inoue, M. Nagura and A. Hirao, Macromol. Res., 2006, 14, 287 Search PubMed.
- Y. Zhao, T. Higashihara, K. Sugiyama and A. Hirao, J. Am. Chem. Soc., 2005, 127, 14158 CrossRef CAS.
- Y. Zhao, T. Higashihara, K. Sugiyama and A. Hirao, Macromolecules, 2007, 40, 228 CrossRef CAS.
- A. Mavroudis and N. Hadjichristidis, Macromolecules, 2006, 39, 535 CrossRef CAS.
- X. Wang, J. He and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4818 CrossRef CAS.
- K. Ueda, A. Hirao and S. Nakahama, Macromolecules, 1990, 23, 939 CrossRef CAS.
- A. Karatzas, H. Iatrou, N. Hadjichristidis, K. Inoue, K. Sugiyama and A. Hirao, Biomacromolecules, 2008, 9, 2072 CrossRef CAS.
- A. Hirao, Y. Tokuda, K. Morifuji and A. Hayashi, Macromol. Chem. Phys., 2001, 202, 1606 CrossRef CAS.
- A. Hirao and Y. Tokuda, Macromolecules, 2003, 36, 6081 CrossRef CAS.
- A. Hirao, K. Kawasaki and T. Higashihara, Macromolecules, 2004, 37, 5179 CrossRef CAS.
- N. Haraguchi and A. Hirao, Macromolecules, 2003, 36, 9364 CrossRef CAS.
- T. Higashihara, M. Nagura, K. Inoue, N. Haraguchi and A. Hirao, Macromolecules, 2005, 38, 4577 CrossRef CAS.
- M. Hayashi, K. Kojima and A. Hirao, Macromolecules, 1999, 32, 2425 CrossRef CAS.
- K. Yamaguchi, A. Hirao, K. Suzuki, K. Takenaka, S. Nakahama and N. Yamazaki, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 395 CrossRef CAS.
- T. Hirano, H.-S. Yoo, Y. Ozama, A. A. El-Magd, K. Sugiyama and A. Hirao, J. Inorg. Organomet. Polym., 2010, 20, 445 CAS.
- P. Fragouli, H. Iatrou, N. Hadjichristidis, T. Sakurai, Y. Matsunaga and A. Hirao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6587 CrossRef CAS.
- S. Paraskeva and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 931 CrossRef CAS.
- H. Iatrou, L. Willner, N. Hadjichristidis, A. Halperin and D. Richter, Macromolecules, 1996, 29, 581 CrossRef CAS.
- H. Iatrou, J. W. Mays and N. Hadjichristidis, Macromolecules, 1998, 31, 6697 CrossRef CAS.
- F. L. Beyer, S. P. Gido, C. Büschl, H. Iatrou, D. Uhrig, J. W. Mays, M. Y. Chang, B. A. Garets, N. P. Balsara, N. B. Tan and N. Hadjichristidis, Macromolecules, 2000, 33, 2039 CrossRef CAS.
- T. Higashihara, R. Faust, K. Inoue and A. Hirao, Macromolecules, 2008, 41, 5616 CrossRef CAS.
- A. Nikopoulou, H. Iatrou, D. J. Lohse and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2597 CrossRef CAS.
- A. Hirao, T. Watanabe and R. Kurokawa, Macromolecules, 2009, 42, 3973 CrossRef CAS.
- A. Hirao, K. Murao, T. Watanabe, R. Kurokawa and K. Sugiyama, Macromolecules, 2009, 42, 7820 CrossRef CAS.
- Y. Tsukahara, J. Inoue, Y. Ohota, S. Kohjiya and Y. Okamoto, Polym. J., 1994, 26, 1013 CrossRef CAS.
- V. Heroguez, Y. Gnanou and M. Fontanille, Macromolecules, 1997, 30, 4792.
- K. Nomura, S. Takahashi and Y. Imanishi, Macromolecules, 2001, 34, 4712 CrossRef CAS.
- D. Pantazis, I. Chalari and N. Hadjichristidis, Macromolecules, 2003, 36, 3783 CrossRef CAS.
- A. Deffieux and M. Schappacher, Macromol. Chem. Phys., 1997, 198, 3953 CrossRef.
- S.-W. Ryu and A. Hirao, Macromolecules, 2000, 33, 4765 CrossRef CAS.
- S.-W. Ryu, H. Asada, T. Watanabe and A. Hirao, Macromolecules, 2004, 37, 6291 CrossRef CAS.
- R. I. Spontak and S. D. Smith, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 947 CrossRef CAS.
- L. Wu, E. W. Cochram, T. P. Lodge and F. S. Bates, Macromolecules, 2004, 37, 3360 CrossRef CAS.
- Y. Matsushita, Macromolecules, 2007, 40, 771 CrossRef CAS.
- H. Watanabe, Y. Matsumiya, T. Sawada and T. Iwamoto, Macromolecules, 2007, 40, 6885 CrossRef CAS.
- K. Sugiyama, T. Oie, A. A. El-Magd and A. Hirao, Macromolecules, 2010, 43, 1403 CrossRef CAS.
- Y. Mogi, M. Nomura, H. Kotsuji, K. Ohonishi, Y. Matsushita and I. Noda, Macromolecules, 1999, 27, 6755.
- E. Giebeler and R. Stadler, Macromol. Chem. Phys., 1997, 198, 3815 CrossRef CAS.
- H. Hückstädt, A. Göpfert and V. Abetz, Polymer, 2000, 41, 9089 CrossRef CAS.
- S. Ludwigs, A. Böker, V. Abetz, A. H. E. Müller and G. Krausch, Polymer, 2003, 44, 6815 CrossRef CAS.
- A. Walther, X. André, M. Drechsler, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2007, 129, 6187 CrossRef CAS.
- A. Walther and A. H. E. Müller, Soft Matter, 2008, 4, 663 RSC.
- T. Tsoukatos, A. Avgeropoulos, N. Hadjichristidis, K. Hong and J. W. Mays, Macromolecules, 2002, 35, 7928 CrossRef CAS.
- T. Ishizone, K. Sugiyama, Y. Sakano, H. Mori, A. Hirao and S. Nakahama, Polym. J., 1999, 31, 983 CrossRef CAS.
- Y. Tanaka, H. Hasegawa, T. Hashimoto, A. Ribbe, K. Sugiyama, A. Hirao and S. Nakahama, Polym. J., 1999, 31, 989 CrossRef CAS.
- E. Giebeler and R. Stadler, Macromol. Chem. Phys., 1997, 198, 3815 CrossRef CAS.
- N. Ekizoglou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1198 CrossRef CAS.
- K. Takahashi, H. Hasegawa, T. Hashimoto, V. Bellas, H. Iatrou and N. Hadjichristidis, Macromolecules, 2002, 35, 4859 CrossRef CAS.
- N. Ekizoglou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2166 CrossRef CAS.
- P. G. Fragouli, H. Iatrou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 514 CrossRef CAS.
- P. G. Fragouli, H. Iatrou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 514 CrossRef CAS.
- P. G. Fragouli, H. Iatrou, D. J. Lohse and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3938 CrossRef CAS.
- T. Oie, Y. Matsuo, K. Sugiyama and A. Hirao, presented in part at the 59th Japan Polymer Society Meeting, Sapporo, Japan, September, 2010.
| This journal is © The Royal Society of Chemistry 2011 |