Synthesis and physical gelation induced by self-assembly of well-defined poly(arylene ether sulfone)s with various numbers of arms†
Jeyoung
Park
,
Myungeun
Seo‡
,
Hyungsam
Choi
and
Sang Youl
Kim
*
Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), 373-1, Guseong-dong, Yuseong-gu, Daejeon, 305-701, Korea. E-mail: kimsy@kaist.ac.kr; Fax: +82 42-350-8177; Tel: +82 42-350-2834
First published on 8th March 2011
Abstract
A series of well-defined poly(arylene ether sulfone)s were synthesized (1P–4P) by chain-growthcondensation polymerization with amideinitiators having various numbers of initiating sites. Differential Scanning Calorimetry (DSC) study of the polymers revealed that branched polymers (3P and 4P) had higher glass transition temperatures (Tg) than linear polymers (1P and 2P) when they had the identical concentration of end groups. However, the viscosity of the polymers decreases as the number of branches increase due to the change of the hydrodynamic volume. Interestingly physical gelation of THF solution of these polymers was observed. Analyses of the self-assembled structure by FE-SEM, FE-TEM, temperature-dependent 1H NMR, FT-IR, and XRD indicated that the formation of fibrillar network was driven by the hydrogen bonding of aromatic amides.
Introduction
Control of polymer architecture is very important to make polymers with controlled properties.1–8 Star-shaped polymers, for example, have a number of arms that are joined at the core and possess different characteristics compared to linear polymers such as smaller hydrodynamic volume and reduced viscosity because of their compact nature.9–11 For the synthesis of well-defined star-shaped polymers with a controlled number and length of arms, use of controlled polymerization methods is essential and controlled polymerization with a multifunctional initiator has been used.1,9–15 However, most of star-shaped polymers synthesized are aliphatic polymers mainly because controlled condensation polymerization of aromatic polymers has been difficult. A recent development of chain-growthcondensation polymerization (CGCP) opens an easy access for well-defined aromatic polymers such as aromatic polyamides and polyethers,16,17 and synthesis of star-shaped aromatic polyamides have been demonstrated.18,19Poly(arylene ethersulfone) (PAES) is one of the well-known high-performance polymers with excellent thermal and mechanical properties,20 but the synthesis of well-defined star-shaped PAES was reported only recently.21 In this contribution, we report the first synthesis of well-defined PAES with various numbers of arms which show that their properties are strongly dependent on the architecture. Interestingly, self-assembly of the well-defined PAESs having amideinitiators induced a physical gelation of the THF solution. Only a few examples of thermoreversible gelation induced by star-shaped polymers have been reported.22–28 This study introduce a new family of polymers which self-assemble and induce a gelation through hydrogen bonds in the core.
Results and discussion
Synthesis of poly(arylene ether sulfone)s from multi-arm initiators
For the synthesis of well-defined PAESsviaCGCP, multifunctional initiators with controlled number of initiating sites were necessary. In this study, four kinds of initiators were synthesized by amidation reaction between 4-fluoro-3-trifluoromethylbenzoylchloride and aromatic amines as shown in Scheme 1. The number of initiating sites was simply controlled by using aromatic compounds with a different number of aminegroups. Because the fluorinegroups in the initiators were activated by the amidecarbonyl group at para position and the trifluoromethyl group at ortho position, the initiators were expected to initiate CGCP of the AB type monomer 1, 4-fluoro-4′-hydroxydiphenyl sulfone potassium salt. | ||
| Scheme 1 Synthesis of multi-arm initiators (1A, 2A, 3A, and 4A). | ||
With the synthesized initiators, polymerization of 4-fluoro-4′-hydroxydiphenyl sulfone potassium salt (1) was carried out according to the reported procedure for linear PAES29 with some modification (Scheme 2). In a typical run, polymerization was conducted in DMSO with 5 wt% monomer concentration using 18-crown-6 as an additive. The feed ratio of monomer to initiator was varied to control the molecular weight of the polymer, and the reaction temperature was maintained at 100 °C to avoid self-polymerization of the monomer. GPC analysis of the resulting polymers in THF indicates that narrow molecular weight distributions (PDI < 1.3) were obtained in every polymerization, suggesting that polymerization proceeded in a controlled manner (Fig. 1).
 | ||
| Fig. 1 THF-GPC profiles of synthesized polymers (RI detector). | ||
The chemical structure of the polymers was analyzed by 1H NMR spectroscopy, which showed that the proton integration ratio of the initiator moiety to the end group is close to 1 as shown in Fig. 2 and S1 (see ESI†). Also, the number average molecular weights (Mn)s of the polymers were obtained by comparing the integral value of the repeating units (A) and the end group (1). The Mns obtained by 1H NMR were smaller than the Mns measured by GPC. The 4-arm star-shaped polymer (4P) has the smallest difference between the Mns determined by 1H NMR and GPC analyses, indicating the polymers possess relatively smaller hydrodynamic volume and elute later during the GPC analysis compared to their linear analogues of similar molecular weight. MALDI-TOF mass spectroscopy analysis (Fig. 3) was conducted to obtain absolute molecular weights of the polymers. The molecular weights measured by MALD-TOF analysis were consistent with Mns based on the 1H NMR analysis. The entire identified mass fraction, separated by the mass of repeating units, contained the mass of the initiator, confirming that PAES with a controlled number and length of arms was synthesized without self-polymerization. Characterization details of the polymers are summarized in Table 1.
 | ||
| Fig. 2 1H NMR spectrum of 1P8.3 (DMSO-d6, 400 MHz). | ||
 | ||
| Fig. 3 MALDI-TOF mass spectra of (a) 1P8.3 and (b) 2P6.3. | ||
| Entry | No. of arms | DP per arm (NMR)a | M n (NMR)a | M n (GPC)b | PDIb | M n (GPC)/Mn (NMR) | T g | [η]d/cm3 g−1 |
|---|---|---|---|---|---|---|---|---|
| a On the basis of the integration of 1H NMR spectra of polymers. b Determined by THF-GPC using polystyrene standards (RI detector). c Measured by DSC with a heating rate of 5 °C min−1 (2nd scan). d Intrinsic viscosity, measured in DMF at 30.5 °C. | ||||||||
| 1P5.7 | 1 | 5.7 | 1610 | 2270 | 1.08 | 1.41 | 151 | 5.4 |
| 1P8.3 | 1 | 8.3 | 2210 | 2950 | 1.18 | 1.33 | 164 | 6.3 |
| 2P5.4 | 2 | 5.4 | 2940 | 4440 | 1.14 | 1.51 | 173 | 10.9 |
| 2P6.3 | 2 | 6.3 | 3400 | 4750 | 1.20 | 1.40 | 177 | 12.0 |
| 3P4.4 | 3 | 4.4 | 3730 | 5550 | 1.15 | 1.49 | 177 | 11.2 |
| 3P5.3 | 3 | 5.3 | 4370 | 5750 | 1.30 | 1.32 | 185 | 12.2 |
| 4P2.9 | 4 | 2.9 | 3580 | 4440 | 1.12 | 1.24 | 165 | 8.6 |
| 4P5.0 | 4 | 5.0 | 5570 | 6740 | 1.26 | 1.21 | 179 | 12.9 |
Thermal properties and viscosities
Thermal properties of the synthesized polymers were studied by DSC. Fig. 4 shows thermograms of the polymers recorded during the 2nd heating cycle at the heating rate of 5 °C min−1 in a nitrogen atmosphere. | ||
| Fig. 4 DSC thermograms of the polymer (2nd heating scan with the heating rate of 5 °C min−1 in N2). | ||
All the polymers exhibited glass transition temperature (Tg) between 150 °C and 180 °C which was strongly dependent on the molecular weight as well as the architecture. According to Ueberreiter and Kanig, the inverse of the Tg can be expressed as a function of the inverse of Mn as shown in eqn (1), considering a polymer as a copolymer of internal units and end groups.30Eqn (1) includes Fox–Flory relation as an approximation limited to higher molecular weight polymers where the weight fraction of the end groups is small.31,32
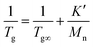 | (1) |
Roovers and Toporowski accounted the effect of the number of arms in the cases of star-shaped polymers by multiplying the number of arms (A) to K′ because the inverse of Mn can be considered as the concentration of the end groups (eqn (2)).33 In Fig. 5, the inverse of Tg is plotted against the number of ends (i.e., end groups) per molecule divided by Mn (NMR), which should be proportional to the volume concentration of end groups in the polymer.
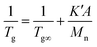 | (2) |
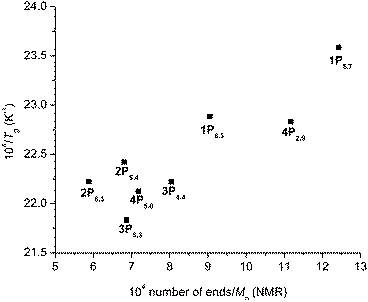 | ||
| Fig. 5 Dependence of glass transition temperature on the number of end groups per molecule divided by the molecular weight of the polymer, i.e., the end group concentration in the sample. | ||
Contradictory to Roovers' results where the identical trend was found for four-/six-arm star-shaped and linear polystyrenes, Fig. 5 showed a distinct difference between the linear polymers (1P and 2P) and the star-shaped polymers (3P and 4P). While 1Ps have the Tg∞ and K′ values same as 2Ps due to the linear architecture, 3Ps and 4Ps have different values. Star-shaped polymers (3P and 4P) possessed higher Tg when they had the identical concentration of end groups (i.e., DP per arm). This effect was attributed to the presence of the central branch point in the star-shaped polymers which encumbers the segmental motion as pointed out by Meares and Bywater.34,35 This architecture-dependent effect was detectable because the polymers used in this study had lower molecular weights compared to Roovers' study and therefore the weight fraction of the central branch points/end groups was significant.
These architectural differences of the polymers also affected the viscosities of polymer solutions. Intrinsic viscosities of the polymer solutions were measured in DMF using Ubbelohde viscometer (Table 1). It is obvious that polymer with higher Mn possessed higher [η] because of larger hydrodynamic volume. However, it is also observed that introduction of a central branch point decreased [η] by reducing the hydrodynamic volume when the molecular weights of the polymers were similar. It can be quantitatively accounted by introducing the branching factor (g′) which is calculated by dividing the intrinsic viscosities of a branched polymer by that of a linear one with the same molecular weight. For the polymers with Mn of ∼3500 g mol−1 in this study, g′ of 3P4.4 was estimated as 0.93 and that of 4P2.9 as 0.72, when 2P6.3 was used as a linear reference. Again, theses values clearly show the effect of the branching architecture on the physical properties of the polymer.
Physical gelation in THF
Conventional poly(arylene ether sulfone)s are usually soluble in methylene chloride and polar aprotic solvents such as NMP and DMF, while insoluble in THF.36 However, the polymers synthesized in this study viaCGCP showed fairly good solubility in THF. Even it was possible to make 10 g L−1 solutions except for 1P8.3, of which the maximum solubility was less than 10 g L−1 even at elevated temperature. However, the transparent solutions in THF (10 g L−1) gradually turned into milky gels at room temperature, except 3P4.4 and 4P2.9 (Fig. 6a). Gelation occurred within 10 minutes in the cases of 1P5.7, 2P5.4, and 2P6.3, while it took several hours for 3P5.3 and 4P5.0. These gels were stable at room temperature but became sol again upon heating. The transition was fully reversible showing no indication of decomposition, after a number of cycles of heating and cooling. | ||
| Fig. 6 (a) Optical image of 1P5.7 gel (left) and 2P5.4 gel (right). FE-SEM images of the xerogel of (b) 1P5.7, (c) 2P5.4, (d) 2P6.3, (e) 3P5.3, (f) 4P5.0, and dried sol of (g) 4P2.9. FE-TEM images of the xerogel of (h) 2P6.3, and (i) a magnified fiber of 2P6.3 in high resolution mode (inset: FFT image). | ||
Field-emission scanning electron microscopy (FE-SEM) showed that the gel was composed of a dense network of fibers with diameters ranging from 10 to 100 nm which immobilized THF inside (Fig. 6b–f), while THF-soluble 3P4.4 and 4P2.9 did not produce such a morphology (Fig. 6g). No notable differences were observed between the polymers having different numbers of arms. Fig. 6h and i show electron micrographs of 2P6.3 gel obtained with a field-emission transmission electron microscope (FE-TEM), indicating the amorphous nature of the fibers.
The solubility of the polymers was tested in other solvents, including n-hexane, ethanol, ethyl acetate, methyl ethyl ketone, acetonitrile, acetone, 1,4-dioxane, methylene chloride, chloroform, ethylene dichloride, toluene, chlorobenzene, and o-dichlorobenzene. As expected, the polymers showed good solubility in chlorinated aliphatic solvents and moderate solubility in acetone but were insoluble in other solvents.
Table 2 summarizes the gelation behavior of the polymer in THF. It turned out that the number of repeating units per arm, not the number of repeating units per polymer, was critical in the formation of gel. The polymers having approximately 5 repeating units per arm formed a gel regardless of the number of arms. The polymers with repeating units less than 5 per arm were soluble in THF and did not form a gel. If the number of repeating units per arm were much greater than 5 (i.e., 1P8.3), the polymer became insoluble in THF like conventional PAES. These solubility properties suggest that the unusual solubility of PAES synthesized in this study originates from the end groups and initiating moiety of the polymers. Because the molecular weights of the polymers are quite low, both the end groups and the initiating moiety occupy significant fractions. Especially the initiating moiety should play an important role since it contains aromatic secondary amides and trifluoromethyl groups which enhance the solubility in THF. Based on the group solubility approximation, relative energy difference (RED) value of PAES in THF was 1.237, indicating that PAES would be insoluble.36 The existence of aromatic secondary amides and trifluoromethyl groups seems to decrease the RED value. If the number of repeating units is less than 5, it is expected that the RED value will be less than 1, making the polymer soluble in THF. As the number of repeating units per arm increases, the relative fraction of the initiating moiety decreases compared to the repeating unit and the enhancement on the solubility in THF should become insignificant.
It seems that the initiating moiety played an important role not only in the enhancement of solubility in THF, but also in the gelation by forming hydrogen bonds between the amides. Temperature-dependent 1H NMR spectroscopy experiments were conducted for 2P6.3 and 4P2.9 in THF-d8 to investigate the role of hydrogen bonding. Fig. 7a illustrates 1H NMR spectra of 2P6.3 in THF-d8 (10 g L−1) recorded at 27 °C (gel state) and 60 °C (sol state). When the temperature increased, the peak corresponding to the amideproton shifted from 9.6 ppm to 9.3 ppm (Δ = 0.27 ppm), indicating the disruption of hydrogen bonds between the amides at elevated temperature. Compared to that, the solution of 4P2.9 in THF-d8 (10 g L−1) maintained its sol state at 27 °C as well as at 60 °C, and the extent of the upfield shift of the peak corresponding to the amideproton was smaller (Δ = 0.17 ppm) than that of 2P6.3 solution.
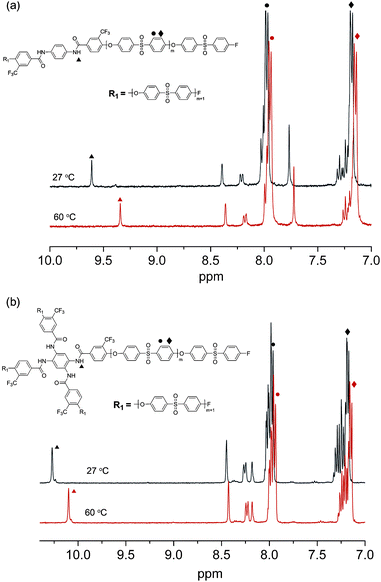 | ||
| Fig. 7 Temperature-dependent 1H NMR spectra of (a) 2P6.3 and (b) 4P2.9 (THF-d8, 400 MHz) at 27, and 60 °C. | ||
FT-IR measurements of the polymer solutions in THF also support the importance of hydrogen bonding in gelation. Fig. 8a shows the amide I and amide II vibrational bands of 2P6.3 in the gel and the sol state, respectively. When the solution became gel upon cooling, the bands showed large red-shift, clearly indicating the formation of hydrogen bonds. The intensity of hydrogen bonding of 3P5.3 was relatively weak reflected in the small red-shift of the amide I and amide II vibrational bands when sol to gel transition occurred (shown in Fig. S2†). The polymer4P2.9 did not show any shift of the amide absorption band as it did not show a sol–gel transition.
 | ||
| Fig. 8 FT-IR spectra of the (a) 2P6.3 and (b) 4P2.9 (10 g L−1, THF). | ||
Taking the evidence together, we propose that gelation of the polymer solution (THF) is mainly driven by the aromatic amide motif at the initiating site which forms hydrogen bonding upon cooling and produces self-assembled nanofibers. It seems that the stability of the gel comes from the effective protection of the hydrogen bonds by surrounded PAES chains which are solvo-phobic in THF, hence prevents THF molecules from the access to the amide. The suggested mechanism resembles to those proposed for low-molecular weight organogelators37–41 rather than polymers,42–45 which can be understood considering well-defined but rather low molecular weight of the polymers used in this study.
Powder X-ray diffraction patterns of the xerogels shown in Fig. 9 also support the above explanation. Because the PAES itself is amorphous,46,47 the as-synthesized 1P5.7 did not show any diffraction. However, xerogel of the polymers showed peaks at the region of 18–30° which can be attributed to the structural periodicity of hydrogen bonded aromatic amides. It is also noted that 1P5.7 and 2P6.3 showed the peaks with relatively higher intensities compared to 3P5.3. It is speculated that the aromatic amides are more crowded as the number of arms connected to the initiator increases and the hydrogen bonds between the amides become weaker. These results are also found in the critical gelation concentration (CGC) of the polymers shown in Table 2, showing higher CGC for polymers with more number of arms.
 | ||
| Fig. 9 Wide-angle powder XRD patterns of the polymers. | ||
Conclusions
Well-defined poly(arylene ether sulfone)s with various numbers of arms were successfully synthesized from multi-arm amideinitiators (1A–4A) via chain-growthcondensation polymerization. The physical properties of the polymers including Tg and viscosity were strongly affected not only by the molecular weight but also by the polymer architecture. Unexpected physical gelation of the polymer solutions attributed to the self-assembly of the polymers induced by the hydrogen bonding between the aromatic amides at the initiating moiety.Acknowledgements
This work was supported by the National Research Foundation (NRF) through NRL (R0A-2008-000-20121-0) program, the Ministry of Environment through Contract No. 20090192091001-B0-0-001-0-0-2009, and Fundamental R&D program for Core Technology of Materials funded by the Ministry of Knowledge Economy, Republic of Korea.References
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS.
- J. Pyun, X. Z. Zhou, E. Drockenmuller and C. J. Hawker, J. Mater. Chem., 2003, 13, 2653–2660 RSC.
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133–1170 CrossRef CAS.
- K. Sada, M. Takeuchi, N. Fujita, M. Numata and S. Shinkai, Chem. Soc. Rev., 2007, 36, 415–435 RSC.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS.
- C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC.
- M. E. Fox, F. C. Szoka and J. M. J. Frechet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS.
- J. Ueda, M. Kamigaito and M. Sawamoto, Macromolecules, 1998, 31, 6762–6768 CrossRef CAS.
- S. Angot, K. S. Murthy, D. Taton and Y. Gnanou, Macromolecules, 1998, 31, 7218–7225 CrossRef CAS.
- E. S. Kim, B. C. Kim and S. H. Kim, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 939–946 CrossRef CAS.
- M. Trollsas and J. L. Hedrick, J. Am. Chem. Soc., 1998, 120, 4644–4651 CrossRef.
- K. Matyjaszewski, Polym. Int., 2003, 52, 1559–1565 CrossRef CAS.
- R. Matmour, A. Lebreton, C. Tsitsilianis, I. Kallitsis, V. Heroguez and Y. Gnanou, Angew. Chem., Int. Ed., 2005, 44, 284–287 CrossRef CAS.
- Z. S. Ge, J. Xu, J. M. Hu, Y. F. Zhang and S. Y. Liu, Soft Matter, 2009, 5, 3932–3939 RSC.
- T. Yokozawa and A. Yokoyama, Prog. Polym. Sci., 2007, 32, 147–172 CrossRef CAS.
- Y. J. Kim, M. Seo and S. Y. Kim, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1049–1057 CrossRef CAS.
- R. Sugi, Y. Hitaka, A. Yokoyama and T. Yokozawa, Macromolecules, 2005, 38, 5526–5531 CrossRef CAS.
- K. Yoshino, A. Yokoyama and T. Yokozawa, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6328–6332 CrossRef CAS.
- J. B. Rose, Polymer, 1974, 15, 456–465 CrossRef CAS.
- J. Park, M. Moon, M. Seo, H. Choi and S. Y. Kim, Macromolecules, 2010, 43, 8304–8313 CrossRef CAS.
- H.-H. Lin and Y.-L. Cheng, Macromolecules, 2001, 34, 3710–3715 CrossRef CAS.
- B. Loppinet, E. Stiakakis, D. Vlassopoulos, G. Fytas and J. Roovers, Macromolecules, 2001, 34, 8216–8223 CrossRef CAS.
- D. Vlassopoulos, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 2931–2941 CrossRef CAS.
- S. J. Lee, B. R. Han, S. Y. Park, D. K. Han and S. C. Kim, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 888–899 CrossRef CAS.
- K. Nagahama, T. Ouchi and Y. Ohya, Adv. Funct. Mater., 2008, 18, 1220–1231 CrossRef CAS.
- F. van de Manakker, T. Vermonden, N. el Morabit, C. F. van Nostrum and W. E. Hennink, Langmuir, 2008, 24, 12559–12567 CrossRef CAS.
- N. Stavrouli, A. Kyriazis and C. Tsitsilianis, Macromol. Chem. Phys., 2008, 209, 2241–2247 CrossRef CAS.
- T. Yokozawa, T. Taniguchi, Y. Suzuki and A. Yokoyama, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3460–3464 CrossRef CAS.
- K. Ueberreiter and G. Kanig, J. Colloid Sci., 1952, 7, 569 CrossRef CAS.
- T. G. Fox and P. J. Flory, J. Appl. Phys., 1950, 21, 581.
- T. G. Fox and P. J. Flory, J. Polym. Sci., 1954, 14, 315 CrossRef CAS.
- J. E. L. Roovers and P. M. Toporowski, J. Appl. Polym. Sci., 1974, 18, 1685–1691 CrossRef CAS.
- P. Meares, in POLYMERS: Structure and Bulk Properties, Van Nostrand Reinhold Company, London, 1965, ch. 10 Search PubMed.
- S. Bywater, Adv. Polym. Sci., 1979, 30, 89–116 CAS.
- C. M. Hansen, in Hansen Solubility Parameters: a User's Handbook, CRC Press, Boca Raton, 2nd edn, 2007, ch. 5 Search PubMed.
- U. Beginn, S. Sheiko and M. Moller, Macromol. Chem. Phys., 2000, 201, 1008–1015 CrossRef CAS.
- J. J. van Gorp, J. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef CAS.
- M. Suzuki, Y. Nakajima, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Langmuir, 2003, 19, 8622–8624 CrossRef CAS.
- C. C. Xue, S. Jin, X. Weng, J. J. Ge, Z. H. Shen, H. Shen, M. J. Graham, K. U. Jeong, H. B. Huang, D. Zhang, M. M. Guo, F. W. Harris, S. Z. D. Cheng, C. Y. Li and L. Zhu, Chem. Mater., 2004, 16, 1014–1025 CrossRef CAS.
- J. Puigmarti-Luis, A. Minoia, A. P. del Pino, G. Ujaque, C. Rovira, A. Lledos, R. Lazzaroni and D. B. Amabilino, Chem.–Eur. J., 2006, 12, 9161–9175 CrossRef CAS.
- S.-k. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- C. Tsitsilianis, Soft Matter, 2010, 6, 2372–2388 RSC.
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2010, 39, 455–463 RSC.
- D. Ka, M. Seo, H. Choi, E. H. Kim and S. Y. Kim, Chem. Commun., 2010, 46, 5722–5724 RSC.
- F. Lufrano, G. Squadrito, A. Patti and E. Passalacqua, J. Appl. Polym. Sci., 2000, 77, 1250–1256 CrossRef CAS.
- R. Guan, H. Zou, D. P. Lu, C. L. Gong and Y. F. Liu, Eur. Polym. J., 2005, 41, 1554–1560 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR and FT-IR spectra of polymers. See DOI: 10.1039/c0py00418a |
| ‡ Current address: Department of Chemistry, University of Minnesota, Minneapolis, MN, 55455-0431, USA. |
| This journal is © The Royal Society of Chemistry 2011 |