High temperature synthesis of vinyl terminated polymers based on dendronized acrylates: a detailed product analysis study†
Anna-Marie
Zorn
a,
Michael
Malkoch
*b,
Anna
Carlmark
b and
Christopher
Barner-Kowollik
*a
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128, Karlsruhe, Germany. Web: www.macroarc.deE-mail: christopher.barner-kowollik@kit.edu; Tel: +49 721 608 45641
bFiber and Polymer Technology, School of Chemical Science and Engineering, Royal Institute of Technology, Teknikringen 56-58, SE-100 44, Stockholm, Sweden. E-mail: malkoch@kth.se; Tel: +46 8 790 8768
First published on 14th March 2011
Abstract
The combination of dendrons and high temperature acrylatepolymerization represents a viable route to form dendronized macromonomers. Dendronized acrylates based on 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) were synthesized using dendrimer synthesis and click chemistry (copper catalyzed azide alkyne cycloaddition (CuAAC)). The synthesis was carried out up to the 3rd generation and with a carbon spacer length of 6 or 9 between the acrylic function and the dendron core. These dendronized acrylates were subjected to auto-initiated high temperature acrylatepolymerization. The polymerization was performed at 140 °C in a 5 wt% solution of hexyl acetate with a 2,2′-azobis(isobutyronitrile) (AIBN) concentration of 5 × 10−3 g mol−1. The vinyl terminated polymers were in-depth characterized viasize exclusion chromatography (SEC) and size exclusion chromatography coupled to electrospray ionization mass spectrometry (SEC-ESI-MS) to assess the generated product spectrum and the efficiency of the process. The achievable number average molecular weight, Mn, was between 1700 and 4400 g mol−1. The degree of polymerization, DPn, decreases with increasing generations of the dendronized acrylates from 6.3 to 3.4. The purity of vinyl terminated oligomers containing a geminal double bond is up to 83%, with the dendronized acrylates of the 1st generation providing the best result. Moderate deprotection of the acetonidegroups occurred spontaneously during the macromonomer formation process and reached its maximum at generation 3.
Introduction
Polymer science focuses on the synthesis of well-designed macromolecules with specific molecular weight, low polydispersity and high endgroup fidelity. In the last decades the interest in dendrimer chemistry rapidly increased due to their monodisperse shape, high branching and the deviant thermal and physical properties of dendritic materials in comparison to linear polymers.1,2 Vögtle et al. first reported on these branched polymeric architectures in the late 1970s using the concept of repetitive growth.3 Subsequently, results in this area were published by Tomalia and colleagues and Newkome et al. followed by the work of Fréchet and co-workers.4,5 Several application areas are conceivable e.g.polymerizationinitiators,6 catalysis,7 liquid crystals,8 molecular encapsulation9 and biomedicine.10Dendritic structures can be divided into several substructures, i.e.dendrimers, hyperbranched polymers, dendrigrafts and dendritic linear hybrids including dendronized polymers.11 The current work addresses dendronized polymers and oligomers, which can be described as linear polymer chains with several dendritic structures in each repeat unit.12 Three main strategies to achieve such structures are described in the literature, i.e. (i) the “grafting-to” approach where premade dendrimers are convergently coupled to functionalized linear polymers, (ii) the “grafting-from” approach where the polymer backbone is stepwise functionalized via divergent growth or (iii) the macromonomer approach where dendrons bearing a vinyl functionality are polymerized. Among these synthesis strategies towards dendronized polymers, the macromonomer approach using radical or controlled/living radical polymerization methods is the most efficient synthetic procedure due to the high levels of control over the dendritic side group incorporation within the macromolecule.13–22
Controlled/living radical polymerization methods such as reversible addition chain transfer (RAFT),23,24 atom transfer radical polymerization (ATRP),25nitroxide-mediated polymerization (NMP)26 as well as ionic processes such as ring-opening polymerization (ROP)27,28 allow for the synthesis of well-defined, almost monodisperse polymers with high endgroup fidelity. However, conventional free radical polymerization techniques can also be employed to obtain polymers with high endgroup fidelity, albeit with higher polydispersity. Chiefari et al. introduced a macromonomer (mm) formation method based on the free radical polymerization of acrylates, which allows obtaining macromonomeric species in a one-pot synthesis.29,30 During polymerization, the formation of macromonomers benefits from the so-called mid-chain radicals (MCR).31–33 These MCR undergo scission reactions at sufficiently high temperatures and low monomer content, resulting in vinyl terminated species.32 The reaction mechanism is well-studied via detailed mass spectrometry and the synthetic approach can be regarded as well established.35,36 Such a direct access route enables the synthesis of highly pure macromonomers via a one-pot–one-step procedure. We recently reported the synthesis of a macromonomer library of acrylates via the above mentioned high temperature acrylatepolymerization method.37 Our interest in simple access routes to vinyl terminated polymers is driven by their applicability as versatile macromolecular building blocks for the assembly of complex polymer designs. The terminal vinylic function can serve as a potentially powerful synthetic handle in variable conjugation chemistries (e.g. Diels–Alder or metathesis).38 In the present contribution, we wish to combine dendrimer chemistry and high temperature acrylatepolymerization to extend the existing macromonomer library.37 Consequently, the present report contains the synthesis of dendronized acrylates based on 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) and their application in high temperature acrylatepolymerization yielding comb-macromonomers. The combination of convergent acetylene bearing dendron synthesis, click chemistry34,39,40 and high temperature acrylatepolymerization is a versatile technique to generate dendronized vinyl terminated polymers. Orthogonal modular ligation appeared to be a highly efficient synthetic pathway to generate the required dendritic acrylate monomers.41,42Scheme 1 depicts the general synthetic strategy followed in the current work.
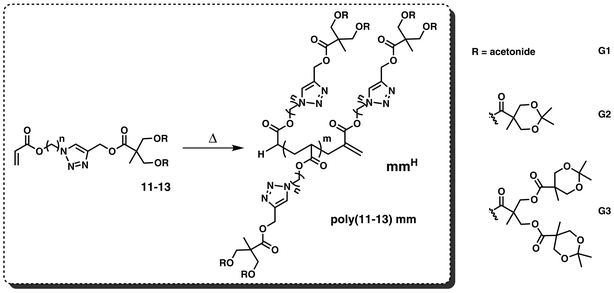 | ||
| Scheme 1 High temperature acrylatepolymerization of dendronized acrylates 11–13 (the numbering is consistent with the compound numbering during the synthesis (see Scheme 2)) forming vinyl terminated polymers/oligomers poly(11–13) mm. Polymerization conditions: hexyl acetate, AIBN, 140 °C, 72 h. | ||
Typically, the performance of a (free radical) polymerization in terms of monomer to polymer conversion decreases with higher generations of dendrons within the monomer due to steric hindrance.18 To avoid this limiting factor a carbon spacer between the polymerizable endgroup and the dendritic structure of the macromonomer is sought to be a tool to improve the polymerization properties.17,18 In the current work a carbon 9 chain (9 –CH2– between the acrylic moiety and the dendritic structure) and a carbon 6 chain are employed, respectively, as a spacing unit. In the literature vinylic dendrons up to generation 4 have been reported to readily polymerize via convenient and controlled radical polymerizations, whereas higher generations are limited in their polymerization efficiency.12
Herein, dendronized acrylates were synthesized up to generation 3 and the subsequent macromonomer formation for all prepared generations was investigated. In addition, the copolymerization of generation 1 dendronized acrylates with ethyl acrylate was explored. In addition, the current study entails the in-depth assessment of the novel dendritic acrylate structures and—most importantly and as one of its core elements—a detailed structural product analysis of the prepared dendritic macromonomers viaSEC as well as (SEC-)ESI-MS, allowing a quantification of the efficiency and viability of the macromonomer formation process.
Experimental part
Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v mixture of THF:methanol with polymer concentration ∼0.2 mg mL−1. The instrumental resolution of the employed experimental set-up is 0.1 amu.
:2 v/v mixture of THF:methanol with polymer concentration ∼0.2 mg mL−1. The instrumental resolution of the employed experimental set-up is 0.1 amu.
For SEC-ESI-MS the LXQ was coupled to a Series 1200 HPLC system (Agilent) that consisted of a solvent degasser (G1322A), a binary pump (G1312A) and a high performance autosampler (G1367B), followed by a thermostatted column compartment (G1316A). Separation was performed on two mixed bed GPC columns (Polymer Laboratories, Mesopore 250 × 4.6 mm, particle dia. 3 μm) with pre-column (Mesopore 50 × 4.6 mm) operating at 30 °C. THF at a flow rate of 0.3 mL min−1 was used as the eluent. The mass spectrometer was coupled to the column in parallel to an RI detector (G1362A with SS420 × A/D) in a setup described previously.44 A 0.27 mL min−1 aliquot of the eluent was directed through the RI detector and 30 μL min−1 infused into the electrospray source after postcolumn addition of a 0.1 mM solution of sodium iodide in methanol at 20 μL min−1 by a micro-flow HPLC syringe pump (Teledyne ISCO, Model 100DM). The polymer solutions (20 μL) with a concentration of 2 mg mL−1 were injected into the HPLC system.
Materials
Hexyl acetate (99%) and palladium on carbon (10% Pd, unreduced) were purchased from Acros and used as received. 2,2′-Azobis(isobutyronitrile) (AIBN) (Sigma–Aldrich, 98%) was recrystallized twice from methanol and ethyl acrylate (Aldrich, 99%) was deinhibited over basic alumina (Acros, Brockmann I, for chromatography, 50–200 μm) prior to use. 9-Bromo-1-nonanol (95%), 6-bromo-1-hexanol (97%), acryloyl chloride (99%), 4-(dimethylamino)pyridine (DMAP) (99%), hydroquinone (99+%) and CuSO4·5H2O (98+%) were purchased from Aldrich and used as received. NaN3 was purchased from Riedel de Haen, propargyl alcohol (99%) from Fluka and (+)-sodium L-ascorbate from Sigma and all used as received. Acetonide-protected 2,2-bis(hydroxymethyl)propionic anhydride (An-bis-MPA) was synthesized according to the literature2 from 2,2-bis(hydroxymethyl)propionic acid (bis-MPA), kindly donated by Perstop AB, Sweden. DOWEX® 50W-X2 50–100 (H) ion-exchange resin (Alfa Aesar) was purified with methanol prior to use. Dimethylsulfoxide (DMSO), triethylamine, dichloromethane (CH2Cl2), methanol and the TLC plates (TLCsilica gel 60 F254) were purchased from Merck and used as received. Pyridine, tetrahydrofuran (THF) and diethylether were all AnalaR Normapur and received from VWR. Ethyl acetate (EtOAc) and n-heptane were p.a. grade and obtained from Fisher Scientific. Column chromatography was performed with Acros Organics silica gel pore size 60 Å, 40–60 μm.Nomenclature
The nomenclature of the dendritic structures and dendronized acrylates is as follows: (R)m-[GX]-acetylene for acetylene terminated dendrons, where R represents the external functional group, e.g. An for acetonide protection and OH for a hydroxyl function with m chain end functionalities. X indicates the generation of the dendritic structure. (R)m-[GX]-triazole-alkyl acrylate (CnGX) represents the corresponding dendronized acrylate with the above described nomenclature. | ||
| Scheme 2 Synthesis of the dendronized acrylates generation 1 to 3. Reaction conditions: (a) NaN3, DMSO, reflux. (b) Acryloyl chloride, NEt3, DMAP, CH2Cl2, rt. (c) DMAP, pyridine, CH2Cl2, rt. (d) DOWEX® H+, methanol, 40 °C. (e) CuSO4·5H2O, sodium ascorbate, THF:H2O (3:1), rt. | ||
For the vinyl terminated polymers/oligomers the following nomenclature applies: the macromonomers mmHnGX-mAn and mmHexnGX-mAn are terminated by a vinyl terminus and a proton or a hexyl side chain and initiated via a transfer to solvent reaction, respectively. Saturated products are labeled with satPnGX-mAn and satPHexnGX-mAn, respectively, with the above mentioned restrictions according to the nomenclature for dendronized acrylates. The subscript of each species represents the amount and nature of the repeat units incorporated in the polymer chain. Thus, the general notation is: n(GX)-m(An) where n represents the respective overall number of repeat units GX lowered by m deprotected acetonide on the dendron surface for the homopolymer and n(GX)-m(An) + ea including the respective number of ethyl acrylate (ea) repeat units for the copolymer (see Scheme S1†).
Synthesis
The dendrons 5–7 have been synthesized according to literature (Scheme 2).456-Azido-1-hexanol (9a). 6-Bromo-1-hexanol 8a (10.0 g, 0.055 mol, 1 eq.) was dissolved in DMSO (100 mL) and heated to 40 °C. Sodium azide (17.9 g, 0.276 mol, 5 eq.) was added stepwise and the reaction mixture was stirred under reflux for 20 h. The organic phase was extracted with ether (3 × 250 mL), dried over MgSO4 and concentrated under reduced pressure. 9a was isolated as a colorless oil (82%).46
1H NMR (400 MHz, CDCl3) δ 3.64 (t, J = 6.5 Hz, 2H, HO-CH2-), 3.27 (t, J = 6.9 Hz, 2H, -CH2-N3), 1.64–1.54 (m, 4H, HO-CH2-CH2-, -CH2-CH2-N3), 1.43–1.34 (m, 4H, -(CH2)2-CH2-CH2-N3). 13C NMR (100 MHz, CDCl3) δ 62.90 (1C, HO-CH2-), 51.51 (1C, -CH2-N3), 32.68 (1C, -CH2-CH2-N3), 28.94 (1C, -CH2-CH2-CH2-N3), 26.65 (1C, HO-CH2-CH2-CH2), 25.46 (1C, HO-CH2-CH2-).
9-Azido-1-nonanol (9b). 9-Azido-1-nonanol was prepared from 9-bromo-1-nonanol8b according to the general procedure for the synthesis of alkyl azides by nucleophilic substitution to give 9b as a colorless oil (94%).
1H NMR (400 MHz, CDCl3) δ 3.63 (t, J = 6.6 Hz, 2H, HO-CH2-), 3.25 (t, J = 7.0 Hz, 2H, -CH2-N3), 1.63–1.52 (m, 4H, HO-CH2-CH2-, -CH2-CH2-N3), 1.33 (d, J = 15.7 Hz, 10H, -(CH2)5-CH2-CH2-N3). 13C NMR (100 MHz, CDCl3) δ 63.20 (1C, HO-CH2-), 51.62 (1C, -CH2-N3), 32.91 (1C, HO-CH2-CH2-), 29.51 (1C, -CH2-CH2-N3), 29.22 (2C, HO-(CH2)3-(CH2)2-), 28.97 (1C, -CH2-(CH2)3-N3), 26.84 (1C, -CH2-(CH2)2-N3), 25.84 (1C, HO-(CH2)2-CH2-).
6-Azido-1-hexyl acrylate (10a). 6-Azido-1-hexanol 9a (6.0 g, 0.042 mol, 1.0 eq.), NEt3 (6.4 g, 0.063 mol, 1.5 eq.), DMAP (1.0 g, 0.008 mol, 0.2 eq.) and hydroquinone (100 ppm) were dissolved in DCM (120 mL) and stirred at ambient temperature. Acryloyl chloride (5.7 g, 0.063 mol, 1.5 eq.) was slowly added to the round bottom flask. The reaction mixture was stirred for 24 h. The solution was diluted with DCM (250 mL) and extracted with 10% NaHSO4 (3 × 200 mL), 10% NaHCO3 (3 × 200 mL) and brine (3 × 200 mL). The organic phase was dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel with ethyl acetate–heptane (v/v 3/7) to give 10a as a colorless oil (81%).
1H NMR (400 MHz, CDCl3) δ 6.40 (dd, J = 17.3, 1.4 Hz, 1H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH-), 6.12 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.82 (dd, J = 10.4, 1.5 Hz, 1H, CH2CH-), 4.16 (t, J = 6.6 Hz, 2H, -CH2-OCO-), 3.27 (t, J = 6.9 Hz, 2H, -CH2-N3), 1.72–1.67 (m, 2H, -CH2-CH2-OCO-), 1.62 (dt, J = 10.9, 7.7 Hz, 2H, -CH2-CH2-N3), 1.42 (dt, J = 7.2, 3.5 Hz, 4H, -(CH2)2-CH2-CH2-N3).
CH-), 6.12 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.82 (dd, J = 10.4, 1.5 Hz, 1H, CH2CH-), 4.16 (t, J = 6.6 Hz, 2H, -CH2-OCO-), 3.27 (t, J = 6.9 Hz, 2H, -CH2-N3), 1.72–1.67 (m, 2H, -CH2-CH2-OCO-), 1.62 (dt, J = 10.9, 7.7 Hz, 2H, -CH2-CH2-N3), 1.42 (dt, J = 7.2, 3.5 Hz, 4H, -(CH2)2-CH2-CH2-N3).
9-Azido-1-nonanyl acrylate (10b). 9-Azido-1-nonanyl acrylate was prepared from 9-azido-1-nonanol9b according to the general esterification procedure for acrylate preparation to give 10b as a colorless oil (80%).
1H NMR (400 MHz, CDCl3) δ 6.39 (dd, J = 17.3, 1.4 Hz, 1H, CH2CH-), 6.12 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.81 (dd, J = 10.4, 1.4 Hz, 1H, CH2CH-), 4.14 (t, J = 6.7 Hz, 2H, -CH2-OCO-), 3.25 (t, J = 6.9 Hz, 2H, -CH2-N3), 1.62 (ddd, J = 14.4, 12.7, 7.2 Hz, 4H, HO-CH2-CH2-, -CH2-CH2-N3), 1.33 (d, J = 17.0 Hz, 10H, -(CH2)5-CH2-CH2-N3).
(An)1-[G1]-triazole-hexyl acrylate (11a). (An)1-[G1]-acetylene 5 (3.0 g, 0.014 mol, 1.1 eq.) and 6-azido-1-hexyl acrylate10a (2.5 g, 0.013 mol, 1.0 eq.) were dissolved in 50 mL THF–H2O (3
:1). CuSO4·5H2O (0.3 g, 1.270 mmol, 0.1 eq.) and sodium ascorbate (1.7 g, 6.340 mmol, 0.5 eq.) were added to the reaction mixture and stirred overnight at ambient temperature. The solvents were evaporated under reduced pressure and the crude product was purified by column chromatography starting with ethyl acetate–heptane (v/v 2/8) and gradually increasing the polarity. The product was isolated as a colorless oil (86%).
1H NMR (400 MHz, CDCl3) δ 7.60 (s, 1H, -N-CHC), 6.39 (dd, J = 17.3, 1.4 Hz, 1H, CH2CH-), 6.11 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.82 (dd, J = 10.4, 1.4 Hz, 1H, CH2CH-), 5.30 (s, 2H, -CH2-OCO-), 4.34 (t, J = 7.2 Hz, 2H, -CH2-N), 4.18 (d, J = 11.9 Hz, 2H, -C-CH2-O- 1st generation), 4.13 (t, J = 6.6 Hz, 2H, O-CH2-), 3.63 (d, J = 11.9 Hz, 2H, -C-CH2-O- 1st generation), 1.96–1.87 (m, 2H, -CH2-CH2-N), 1.70–1.61 (m, 2H, O-CH2-CH2-), 1.38 (d, J = 27.6 Hz, 10H, C(CH3)2, -(CH2)2-), 1.13 (s, 3H, C-CH3 1st generation). 13C NMR (100 MHz, CDCl3) δ 174.34 (1C, -CH2OCO-), 166.40 (1C, H2CC-CO-), 143.07 (1C, N-CHC), 130.83 (1C, H2CCH-), 128.60 (1C, H2CCH-), 123.59 (1C, N-CHC), 98.21 (1C, C(CH3)2), 66.12 (2C, C-(CH2O)2), 64.37 (1C, -CH2-OCO-), 58.35 (1C, -CHC-CH2-), 50.39 (1C, -CH2N), 42.06 (1C, -OCO-C-), 30.27 (1C, -O-CH2-CH2), 28.52 (1C, -CH2-CH2-N), 26.26 (1C, -CH2-(CH2)2-N), 25.53 (1C, C(CH3)2), 25.31 (1C, C(CH3)2), 22.24 (1C, -O-(CH2)-CH2-), 18.55 (1C, -OCO-CCH3). ESI-MS[MNa]+ theo. 432.21 Da, exp. 432.25 Da.
(An)1-[G1]-triazole-nonanyl acrylate (11b). (An)1-[G1]-triazole-nonanyl acrylate was synthesized from (An)1-[G1]-acetylene5 and 9-azido-1-nonanyl acrylate10b according to the general procedure for the copper catalyzed azide alkyne cycloaddition (CuAAC) to give 11b as a colorless oil (75%).
1H NMR (400 MHz, CDCl3) δ 7.60 (s, 1H, -N-CHC), 6.39 (dd, J = 17.3, 1.4 Hz, 1H, CH2CH-), 6.11 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.81 (dd, J = 10.4, 1.4 Hz, 1H, CH2CH-), 5.30 (s, 2H, -CH2-OCO-), 4.32 (t, J = 7.3 Hz, 2H, -CH2-N), 4.18 (d, J = 11.8 Hz, 2H, C-CH2-O- 1st generation), 4.13 (t, J = 6.7 Hz, 2H, -O-CH2-), 3.63 (d, J = 11.8 Hz, 2H, -C-CH2-O- 1st generation), 1.94–1.81 (m, 2H, -CH2-CH2-N), 1.64 (dd, J = 14.3, 6.9 Hz, 2H, -O-CH2-CH2-), 1.42 (s, 3H, C(CH3)2), 1.35 (s, 3H, C(CH3)2), 1.29 (d, J = 3.8 Hz, 10H, -(CH2)5-), 1.13 (s, 3H, C-CH3 1st generation). 13C NMR (100 MHz, CDCl3) δ 174.33, 166.49, 142.98, 130.66, 128.72, 123.59, 98.22, 66.11, 64.75, 58.34, 50.53, 42.05, 30.35, 29.28, 29.00, 28.68, 26.55, 25.97, 25.31, 22.23, 18.54. ESI-MS [M + Na]+ theo. 474.26 Da, exp. 474.33 Da.
(An)2-[G2]-triazole-hexyl acrylate (12a). (An)2-[G2]-triazole-hexyl acrylate was synthesized from (An)2-[G2]-acetylene6 and 6-azido-1-hexyl acrylate10a according to the general procedure for the copper catalyzed azide alkyne cycloaddition (CuAAC) to give 12a as a colorless oil (75%).
1H NMR (400 MHz, CDCl3) δ 7.62 (s, 1H, N-CHC), 6.39 (dd, J = 17.3, 1.4 Hz, 1H, CH2CH-), 6.11 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.82 (dd, J = 10.4, 1.4 Hz, 1H, CH2CH-), 5.26 (s, 2H, -CH2-OCO-), 4.34 (dd, J = 4.3, 2.9 Hz, 2H, -CH2-N), 4.31 (s, 4H, -C-CH2-O- 1st generation), 4.15 (d, J = 6.6 Hz, 2H, -O-CH2-), 4.13–4.07 (m, 4H, -C-CH2-O- 2nd generation), 3.58 (d, J = 12.2 Hz, 4H, -C-CH2-O- 2nd generation), 1.98–1.87 (m, 2H, -CH2-CH2-N-), 1.74–1.62 (m, 2H, -O-CH2-CH2-), 1.41 (s, 6H, C(CH3)2 2nd generation), 1.39 (s, 4H, -(CH2)2-), 1.34 (s, 6H, C(CH3)2 2nd generation), 1.27 (s, 3H, C-CH3 1st generation), 1.10 (s, 6H, C-CH3 2nd generation). 13C NMR (100 MHz, CDCl3) δ 173.64, 166.42, 130.85, 128.60, 123.90, 116.29, 98.25, 66.07, 65.36, 64.39, 58.71, 50.41, 46.95, 42.16, 30.27, 28.54, 26.32, 25.49, 22.12, 18.61. ESI-MS [M + Na]+ theo. 704.34 Da, exp. 704.42 Da.
(An)2-[G2]-triazole-nonanyl acrylate (12b). (An)2-[G2]-triazole-nonanyl acrylate (12b) was synthesized from (An)2-[G2]-acetylene6 and 9-azido-1-nonanyl acrylate10b according to the general procedure for the copper catalyzed azide alkyne cycloaddition (CuAAC) to give 12b as a colorless oil (65%).
1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H, N-CHC), 6.39 (dd, J = 17.3, 1.4 Hz, 1H, CH2CH-), 6.12 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.82 (dd, J = 10.4, 1.4 Hz, 1H, CH2CH-), 5.26 (s, 2H, -CH2-OCO-), 4.33 (d, J = 7.6 Hz, 2H, -CH2-N), 4.31 (s, 4H, C-CH2-O- 1st generation), 4.15 (d, J = 6.7 Hz, 2H, O-CH2-), 4.12–4.07 (m, 4H, -C-CH2-O- 2nd generation), 3.58 (d, J = 12.2 Hz, 4H, -C-CH2-O- 2nd generation), 1.94–1.86 (m, 2H, -CH2-CH2-N), 1.70–1.62 (m, 2H, O-CH2-CH2-), 1.41 (s, 6H, C-CH3 2nd generation), 1.34 (s, 16H, C(CH3)2 2nd generation, -(CH2)5-), 1.27 (s, 3H, C-CH3 1st generation), 1.10 (s, 6H, C(CH3)2 2nd generation). 13C NMR (100 MHz, CDCl3) δ 173.63, 172.70, 142.43, 130.67, 128.72, 123.87, 98.24, 66.06, 65.36, 64.75, 58.72, 50.54, 46.94, 42.15, 30.37, 29.39, 29.04, 28.70, 26.62, 25.99, 25.42, 22.11, 18.60, 17.80. ESI-MS [M + Na]+ theo. 746.38 Da, exp. 746.36 Da.
(An)4-[G3]-triazole-hexyl acrylate (13a). (An)4-[G3]-triazole-hexyl acrylate (13a) was synthesized from (An)4-[G3]-acetylene7 and 6-azido-1-hexyl acrylate10a according to the general procedure for the copper catalyzed azide alkyne cycloaddition (CuAAC) to give 13a as a colorless oil (50%).
1H NMR (400 MHz, CDCl3) δ 7.72 (s, 1H, -N-CHC), 6.39 (dd, J = 17.3, 1.4 Hz, 1H, CH2CH-), 6.11 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.82 (dd, J = 10.4, 1.4 Hz, 1H, CH2CH-), 5.26 (s, 2H, CH2-OCO), 4.37 (t, J = 7.3 Hz, 2H, -CH2-N), 4.29–4.24 (m, 10H, C-CH2-O- 2nd generation, O-CH2-), 4.23 (d, J = 4.0 Hz, 2H, C-CH2-O- 1st generation), 4.17–4.11 (m, 10H, C-CH2-O- 3rd generation, 1st generation), 3.62 (d, J = 12.2 Hz, 8H, C-CH2-O- 3rd generation), 1.98–1.87 (m, 2H, -CH2-CH2-N), 1.71–1.62 (m, 6H, O-CH2-(CH2)3-), 1.41 (s, 12H, C(CH3)2 3rd generation), 1.34 (s, 12H, C(CH3)2 3rd generation), 1.24 (d, J = 2.7 Hz, 9H, C-CH3 1st generation, 2nd generation), 1.13 (s, 12H, C-CH3 3rd generation). 13C NMR (100 MHz, CDCl3) δ 173.69, 171.94, 130.85, 128.61, 116.29, 98.27, 77.48, 77.16, 76.85, 66.10, 65.04, 46.97, 42.21, 25.57, 21.97, 18.62, 17.78. ESI-MS [M + Na]+ theo. 1248.59 Da, exp. 1248.56 Da. MALDI-MS [M + Na]+ theo. 1248.59 Da, exp. 1248.87 Da.
(An)4-[G3]-triazole-nonanyl acrylate (13b). (An)4-[G3]-triazole-nonanyl acrylate (13b) was synthesized from (An)4-[G3]-acetylene7 and 9-azido-1-nonanyl acrylate10b according to the general procedure for the copper catalyzed azide alkyne cycloaddition (CuAAC) to give 13b as a colorless oil (70%).
1H NMR (400 MHz, CDCl3) δ 7.71 (s, 1H, -N-CHC), 6.39 (dd, J = 17.3, 1.5 Hz, 1H, CH2CH-), 6.11 (dd, J = 17.3, 10.4 Hz, 1H, CH2CH-), 5.81 (dd, J = 10.4, 1.5 Hz, 1H, CH2CH-), 5.26 (s, 2H, -CH2-OCO-), 4.35 (t, J = 7.3 Hz, 2H, -CH2-N), 4.31–4.18 (m, 12H, -C-CH2-O- 1st generation, 2nd generation), 4.17–4.11 (m, 10H, O-CH2-, -C-CH2-O- 3rd generation), 3.62 (d, J = 12.2 Hz, 8H, -C-CH2-O- 3rd generation), 1.95–1.84 (m, 2H, -CH2-CH2-N), 1.65 (dd, J = 14.3, 7.0 Hz, 2H, -O-CH2-CH2-), 1.41 (s, 12H, C(CH3)2 3rd generation), 1.33 (d, J = 7.2 Hz, 22H, -(CH2)5-, C(CH3)2 3rd generation), 1.24 (d, J = 3.1 Hz, 9H, C-CH3 1st generation, 2nd generation), 1.13 (s, 12H, C-CH3 3rd generation). 13C NMR (100 MHz, CDCl3) δ 173.66, 171.93, 142.01, 130.65, 128.74, 124.31, 98.24, 66.10, 65.02, 64.75, 58.73, 50.52, 46.86, 42.20, 39.43, 30.43, 29.26, 26.62, 26.00, 25.49, 22.04, 18.63, 17.78. ESI-MS [M + Na]+ theo. 1290.64 Da, exp. 1290.64 Da. MALDI-MS [M + Na]+ theo. 1290.64 Da, exp. 1290.94 Da.
Results and discussion
Dendronized acrylates synthesis
The synthesis and subsequent high temperature polymerization of dendronized acrylates to yield dendronized vinylic macromonomers can be a versatile route towards brush-shaped polymers. Since acrylates undergo β-scission during polymerization resulting in MCR,32 it is possible to generate macromonomers, which can be used in further transformations due to their vinyl terminus. For the synthesis of the dendronized monomers acetylene-functionalized dendrons based on bis-MPA were synthesized according to Hawker and co-workers.45 Dendrons, from 1st to 3rd generation, were accomplished by a divergent growth approach, which combines esterification steps using acetonide protected bis-MPA anhydride and deprotection (activation) with DOWEX® resin. The acetylene cores were subsequently coupled to antecedently synthesized azido-functionalized acrylates via the copper catalyzed azide alkyne cycloaddition (CuAAC). The ligation reaction was catalyzed by copper(I)sulfate in the presence of sodium ascorbate yielding pure dendronized acrylates with a 1,4-disubstituted 1,2,3-triazole linkage after column chromatographic purification. Scheme 2 depicts a detailed overview of the synthetic strategy including the synthesis of acetylene-terminated dendrons, azido-functionalized acrylates and the copper catalyzed azide alkyne cycloaddition (CuAAC) click reaction.To enable an as efficient as possible polymerization/oligomerization, i.e. macromonomer formation process,37 a flexible spacer was introduced into the monomer between the dendron and the acrylic moiety to increase the spatial availability of the acrylic group. Two carbon spacers were introduced containing 6 and 9 CH2 units, respectively. For the azide terminated acrylates 6-bromo-hexanol8a and 9-bromo-nonanol8b, respectively, were reacted with NaN3 including a bromine–azide replacement followed by an esterification step with acryloyl chloride. After column chromatography with heptane:ethyl acetate, the acrylate precursors were isolated with yields of 81% and 80%, respectively.
In the following modular ligation step, the dendronized acrylates were isolated in good yields up to 89% after column chromatography. To prevent the premature polymerization of the generated monomers hydroquinone was added to all reactions, as well as during storage of these compounds. The dendronized acrylates were characterized viaNMR spectroscopy and mass spectrometry as well as ESI and MALDI-TOF.
Macromonomer formation via high temperature acrylatepolymerization
In a significant extension of the previously synthesized macromonomer library,37 the prepared dendronized acrylates were successfully polymerized via high temperature polymerization to afford vinyl terminated oligomeric building blocks. The dendronized acrylates from generation 1 to generation 3, with a spacer length of 6 and 9 each (11–13), were deinhibited over neutral alumina prior to use. Auto-initiated high temperature acrylatepolymerization was carried out in hexyl acetate and small amounts of AIBN were added prior to polymerization.36,37 It is hypothesized that AIBN does not initiate any polymerization activity as all initiator is decomposed within 0.15 minutes at 140 °C.37,47 However, it removes minimal impurities in the system to generate products with high purity. Scheme 1 represents the general idea of the comb-like macromonomer formation based on dendronized acrylates from the 1st to 3rd generation.In the current study, the high temperature polymerization of the 1st generation, the 2nd generation and the 3rd generation dendron with a carbon spacer 6 and 9 each (11–13) was carried out. Below, the analytical process and results will be discussed on the example of the 1st generation with the carbon 9 spacer (11b). The corresponding analysis for higher generations and spacer lengths can be found in the ESI†. The successful polymerization process was initially evidenced by size exclusion chromatography. Fig. 1 presents the SEC chromatogram obtained of p(11b) mm. SEC chromatograms of p(11–13) mm can be found in the ESI† (Fig. S5, p(11a) mm; Fig. S7, p(11b)-co-ea mm; Fig. S9, p(12a) mm; Fig. S11, p(13a) mm; Fig. S13, p(13b) mm).
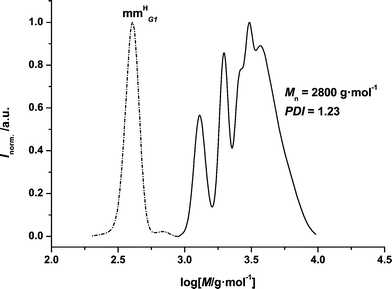 | ||
| Fig. 1 SEC chromatogram of p(11b) mm. The trace on the left hand side represents the residual monomer, whereas the trace on the right hand side shows the distribution after the polymerization. | ||
Fig. 1 displays two distributions: on the left hand side (dashed signal) the residual dendron can be seen. On the right hand side, the distribution of the macromonomer species is depicted. Both signals have been analysed and normalized separately against polystyrene standards. Due to the relatively high molecular weight of the monomer (M(11b) = 451.27 g mol−1), the product distribution on the right hand side reflects the stepwise addition of the monomer to the growing polymer chain with well-resolved peaks. The system never reaches full conversion even at extended reaction times. Therefore a standard reaction time of 72 h for each polymerization was selected. Nevertheless, it is notable that the dendronized acrylates form macromonomers despite their bulky side chains, even for the 3rd generation species. With increasing dendron size, the formation of the macromonomers is limited to smaller numbers of repeat units in the final polymer. However, even in the 3rd generation material, the vinyl terminus is still detectable.
For p(11b) mm the number average molecular weight is close to Mn = 2800 g mol−1, which translates to a degree of polymerization of DP = 6.3. The corresponding p(13b) mm (M(13b) = 1267.65 g mol−1) shows a degree of polymerization of DP = 3.4 at a molecular weight of Mn = 4300 g mol−1 calculated from SEC. The tendency of the dendronized acrylates to form macromonomers with a large polymer backbone decreases with increasing dendritic generation of the monomer—as expected—due to the increasing steric hindrance during the polymerization process.
While the SEC analysis provides an indication that the dendronized acrylates polymerize indeed, a more detailed molecular proof is required. Thus, ESI-MS spectra were measured for all samples to analyse the macromonomers with respect to their endgroup fidelity and purity.48,49 The molecular weights of the generation 1 and 2 macromonomers are well in the range of the calibrated m/z range of the ESI-MS experimental set-up, while the macromonomers of generation 3 were analysed via a hyphenated SEC-ESI-MS due to the high molecular weight of the monomer (M(13a) = 1225.60 g mol−1, M(13b) = 1267.65 g mol−1). Such a slice by slice ESI-MS analysis allows for a better ionization of higher molecular weight polymers in higher charge states, thus allowing an improved imaging of the material.
The synthetic procedure yields macromonomer species of the general structure as detailed in our previous study on acrylate-dented macromonomers.37 During the polymerization one major product is formed, however, in general saturated and unsaturated species can be distinguished. The unsaturated products—the main synthetic target—with their vinyl terminus are capable of undergoing further transformations to construct more complex macromolecules. The unsaturated species mmH represents the main product of the synthesis; a separate species initiated via a transfer to solvent reaction, thus carrying a hexyl acetate radical fragment (mmHex), is also identified. Both species carry a vinyl terminus and are thus equivalent in terms of their chemical reactivity. The saturated species satP and satPHex are only formed in small quantities.
Fig. 2 depicts the ESI-MSspectrum of p(11b) mm as an example. Recall that the ESI-MSspectrum provides a number distribution. The spectrum represents the overall region from 400–3500 m/z where all signals associated with the main product mmHG1 are assigned. The species mmHG1 is related to the residual monomer found in the sample and species mmHnG1 refers to increasing addition of monomers with increasing n. In Fig. 3 a zoom spectrum of a repeat unit for a detailed assignment of all detected species is shown. The experimental and theoretical m/z are summarized in Table S1 (refer to the ESI†) for all detected species. All mass assignments discussed in the following are accurate within ±0.3 Da.
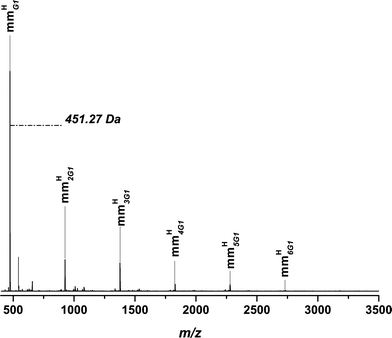 | ||
| Fig. 2 Overall ESI-MSspectrum of p(11b) mm. The polymer was synthesized via high temperature acrylatepolymerization in a solution of hexyl acetate with 5 wt% monomer at 140 °C. | ||
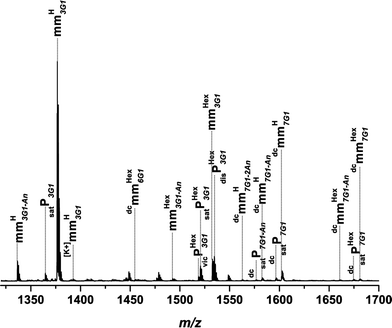 | ||
| Fig. 3 ESI-MS spectrum of p(11b) mm. A zoom spectrum of one repeat unit (451.27 Da) is depicted. The polymer was synthesized via high temperature acrylatepolymerization in a solution of hexyl acetate with 5 wt% monomer at 140 °C. | ||
Inspection of Fig. 3 shows as the main product the mmHnG1 species as well as mmHexnG1 in smaller quantities. The prefix to G1 represents the number of repeat units in the vinyl terminated polymer/oligomer. It should be noted that deprotection of the acetonide decorated surface of the dendron side chain takes place during the polymerization process (−40.03 Da for the acetonide protection in a single charged species). Fortunately, the deprotection (mmHnG1-An) occurs only in small amounts and since the deprotected species are still capable of undergoing further transformations due to their vinylic terminus, it is even conceivable to additionally use the deprotected hydroxyl functions for further transformation such as esterifications. Saturated products satP and satPHex are formed in very minor amounts. In the zoom spectrum, corresponding to a repeat unit of 3 in the polymer, double charged species are detectable, which evidence a propagation of the monomer to up to 7 repeat units. As mentioned above, with increasing generation of the dendron segment in the monomer the propagation ability of the monomer decreases resulting in a lower degree of polymerization.
To unambiguously evidence the vinyl terminus, hydrogenation of the sample was carried out. Fig. 4 shows the zoom spectrum of p(11b) mm and its hydrogenated counterpart.
 | ||
| Fig. 4 ESI-MS spectra of the vinyl terminated p(11b) mm (top) versus the hydrogenated polymer (bottom). | ||
As can be clearly seen in Fig. 4, the vinyl terminated species mmH3G1, mmH3G1-An and dcmmH6G1-An shift by 2 Da (1 Da for double charged species) to higher molecular weight, while all saturated species as satP3G1, dc satP6G1 and dc satP3G1-An are not shifted. Thus, the chemical nature of the macromonomer species is fully established.
For dendron generations 2 and 3, similar product distributions can be found in the mass spectra. Further (SEC-)ESI-MS spectra can be found in the ESI† (Fig. S6, p(11a) mm; Fig. S8, p(11b)-co-ea mm; Fig. S10, p(12a) mm; Fig. S12, p(13a) mm; Fig. S14, p(13b) mm). Table 1 shows an overview of the macromonomer content after each polymerization resulting from calculations of the measured ESI-MS spectra. Calculations were carried out analogous to the method used for the previously synthesized macromonomer library.37
| mma (%) | mmH (%) | An-protectedc (%) | mmHd (%) | M n/g mol−1 | |
|---|---|---|---|---|---|
| a , b Amount of vinyl terminated polymer, containing b proton terminated macromonomers mmH. c , d Amount of acetonide-protected species, containing d proton terminated macromonomer mmH. e Values were calculated from SEC-ESI-MS spectra employing one repeating unit. Integration on other retention volumes provides similar results, however, the error associated with the data given for poly(13) mm is significantly higher than for the other macromonomers. | |||||
| Poly(11a) mm | 83.2 | 68.5 | 65.1 | 60.3 | 3000 |
| Poly(11a)-co-ea mm | 59.5 | 77.9 | 50.5 | 77.4 | 1900 |
| Poly(11b) mm | 81.7 | 91.4 | 82.1 | 83.5 | 2800 |
| Poly(11b)-co-ea mm | 69.9 | 77.7 | 66.5 | 81.1 | 1700 |
| Poly(12a) mm | 70.3 | 92.5 | 85.9 | 76.8 | 2900 |
| Poly(13a) mme | 54.4 | 74.4 | 8.7 | 53.63 | 4400 |
| Poly(13b) mme | 58.5 | 50.9 | 18.0 | 52.7 | 4300 |
Column 1 depicts the content of macromonomer species with a vinyl terminus in the sample. Column 2 contains vinyl terminated polymers with a proton terminus mmH related to the overall amount of macromonomeric species with a vinyl terminus. Vinyl terminated polymers as useful synthetic building blocks are built in acceptable yields exceeding 70%, except for 3rd generation macromonomeric products. The values for poly(13) mm were calculated from SEC-ESI-MS spectra (refer to the provided retention time of the sample shown in Fig. S12 and S14†).
In each sample, the mmH species occurs as the main product and all mentioned side products are only detectable in small quantities. However, it has to be noted that the SEC-ESI-MS spectra for the 3rd generation are slightly deviant from the ESI-MS spectra of the lower generations. Due to the higher sensitivity of the ESI-MS after the pre-separation viaSEC the side products are better visible for each retention time. However, the overall (low) integral amount of side products is identical between ESI-MS and SEC-ESI-MS. Compared to the 1st and 2nd generation macromonomers, deprotection of the acetonide surface occurs to a slightly higher extent, which affects the purity of protected species during the polymerization process. For the 3rd generation protected species up to 18% are present, whereas the amount of acetonide protected species is 51% to 86%, depending on generation and spacer length. Even for the species mmHG3, representing the residual monomer, the deprotection of the acetonide surface is higher compared to lower generations. It is assumed that the higher the generation of the dendrons, the higher the deprotection. At high temperature deprotection occurs to a certain extent, i.e. for a 3rd generation acrylate 8 hydroxy functionalities are protected with 4 acetonide (An) protection groups. Thus, 4 deprotections occur—at the maximum—for each repeat unit. The quantification of the deprotected species is beset with some uncertainty since deprotection could occur on each repeat unit in the system. For example, in the mmH3G3-4An macromonomer with 3 repeat units 4 deprotections took place (m/z = 1195.93). These deprotections could have taken place partly on each repeat unit or—alternatively—completely deprotected one entire monomer unit. Note that the deprotected species still carry a vinylic terminus.
While it is pleasing to note that the dendritic acrylates undergo transformations to macromonomer species, it is mandatory to establish whether copolymeric macromonomers can also be generated. For this purpose the copolymerization behaviour of the acrylic dendrons 11 with ethyl acrylate was explored. In the following the copolymerization of 11a with ethyl acrylate will be discussed in detail. The analytical results for the dendronized acrylate11b can be found in the ESI†. A ratio of 1:1 for the comonomers dendritic acrylate (11):ethyl acrylate has been applied in a 5 wt% solution in hexyl acetate as for the homopolymerization. The SEC trace in Fig. 5 shows a clear shift to higher molecular weights, which proves the presence of a polymerization process; the residual monomer is also detectable.
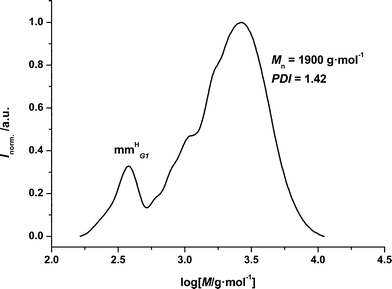 | ||
| Fig. 5 SEC chromatogram of p(11a)-co-ea mm. The peak on the left hand side represents the residual monomer, whereas the shifted part on the right hand side corresponds to the distribution of the copolymerization product. | ||
To establish whether the generated polymeric material is truly a macromonomeric copolymer, a molecular assessment viaESI-MS is required. Fig. 6 shows a typical ESI-MSspectrum of p(11a)-co-ea mm with the overall spectrum from 400 to 2000 m/z (above) and a specific zoom spectrum to a repeat unit of ethyl acrylate (below).
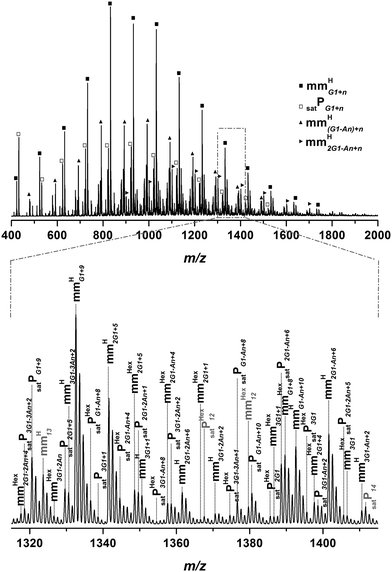 | ||
| Fig. 6 ESI-MS spectrum of p(11a)-co-ea mm. The overall spectrum (above) and the zoom spectrum of a repeat unit (100.05 Da EA) (below) are depicted. The polymer was synthesized via high temperature acrylatepolymerization in a solution of hexyl acetate with 2.5 wt% monomer and 2.5 wt% EA at 140 °C in an oxygen free atmosphere with 5 × 10−3 mol L−1AIBN. | ||
The main distribution in the spectrum can clearly be assigned to mmHG1+n, i.e. the copolymer with one dendronized monomer as repeat unit followed by nethyl acrylaterepeat units (100.05 Da). The second intense distribution corresponds to a deprotected species mmHG1-An+n. The achievable amount of saturated species is higher compared to the high temperature polymerization of pure macromonomer. Since deprotection occurs for the 1st generation acrylate, 3 different monomers acting as separate comonomers are present in the system: protected dendronized acrylate, deprotected dendronized acrylate and ethyl acrylate (see Scheme S2†). This fact leads to a large variety of species found in the ESI-MSspectrum. The calculation of the macromonomer content from ESI-MS measurements indicates a decrease in the macromonomeric species compared to the homopolymerization of the dendronized acrylates. Table 1 indicates a macromonomer content of 83% for poly(11a) mm and 60% for the copolymerpoly(11a)-co-ea mm, respectively. Even in the 2nd generation the formation of vinyl terminated polymers is slightly lower for the copolymerpoly(11b)-co-ea mm (70%) compared to the homopolymerpoly(11b) mm with 82% macromonomer. Since the copolymerization with a small acrylate as a backbone spacer does not result in a more efficient synthesis of well-defined macromonomers with a higher molecular weight, it can be concluded that the copolymerization itself is a versatile route to create molecules with different chemical and physical properties. However, it is recommended to use the dendronized acrylates in the homopolymerization processes to obtain as pure as possible dendronized macromonomers.
The role of the size of the side chain spacer between the acrylic moiety and the dendrons cannot easily be established. Even though the influence of the amount of generations of the dendron on the propagation during the polymerization is clearly evidenced, the carbon spacer between the acrylic moiety and the dendron does not affect the polymerization process. Thus, the spacer length chosen for the monomer synthesis is too similar to impart a significant influence on the polymerization in the high temperature acrylate synthesis of macromonomers.
Conclusions
Dendronized acrylates based on bis-MPA have successfully been synthesized viacopper catalyzed azide alkyne cycloaddition (CuAAC). The benign click reaction gives rise to the combination of an acrylic functionality with well-defined dendritic wedges. The monomers were analyzed viaNMR, ESI-MS and MALDI-TOF.These dendronized acrylates from 1st to 3rd generation, containing a carbon spacer, were subjected to a high temperature acrylate synthesis forming vinyl terminated polymers. For a better availability of the polymerizable group two flexible spacers, carbon 6 and 9, have been introduced into the dendronized monomer structures between the acrylic moiety and the dendronized structures. In addition, the homopolymerization of the molecules and the copolymerization with ethyl acrylate were carried out. The macromonomers were analyzed viaSEC and (SEC-)ESI-MS. The approach of subjecting these monomers to the high temperature acrylate synthesis thus constitutes a feasible avenue for generating versatile synthetic building blocks for further macromolecular transformations.
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG) for the current work is gratefully acknowledged. C.B.-K. acknowledges additional funding for the current project from the Karlsruhe Institute of Technology (KIT) in the context of the Excellence Initiative for leading German universities as well as funding from the Ministry of Science and Arts of the state of Baden-Württemberg. A.-M. Z. and C. B.-K. are grateful for financial support from the Karlsruhe House of Young Scientists (KHYS) enabling a research stay of A.-M. Z. at the KTH. M. M. acknowledges the Swedish Research Council (VR) Grant 2006-3617 for financial support. The authors thank Prof. Eva Malmström (KTH) and Prof. T. Junkers (U Hasselt) for helpful discussions.References
- S. M. Grayson and J. M. J. Frechet, Chem. Rev., 2001, 101, 3819–3867 CrossRef CAS.
- M. Malkoch, E. Malmström and A. Hult, Macromolecules, 2002, 35, 8307–8314 CrossRef CAS.
- (a) E. Buhleier, W. Wehner and F. Vögtle, Synthesis, 1978, 155–158 CrossRef CAS; (b) F. Vogtle, S. Gestermann, R. Hesse, H. Schwierz and B. Windisch, Prog. Polym. Sci., 2000, 25, 987–1041 CrossRef CAS.
- D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719–2728 CrossRef CAS.
- G. R. Newkome, Z. Yao, G. R. Baker and Y. K. Grupta, J. Org. Chem., 1985, 50, 2003–2004 CrossRef CAS.
- M. Trollsås, J. L. Hendrik, D. Meccerreyes, P. Dubois, R. Jerome, H. Ihre and A. Hult, Macromolecules, 1997, 30, 8508–8511 CrossRef CAS.
- J. W. J. Knapen, A. W. van der Made, J. C. de Wilde, A. W. van Leeuwen, P. Wijkens, D. M. Grove and G. van Koten, Nature, 1994, 372, 659–663 CrossRef CAS.
- P. Busson, H. Ihre and A. Hult, J. Am. Chem. Soc., 1998, 120, 9070–9071 CrossRef CAS.
- J. F. G. A. Jansen, E. M. M. de Brabander-Van den Berg and E. W. Meijer, Science, 1994, 266, 1226–1229 CrossRef CAS.
- A. Cordova and K. D. Janda, J. Am. Chem. Soc., 2001, 123, 8248–8259 CrossRef CAS.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- A. D. Schlüter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864 CrossRef CAS.
- W. Li, D. Wu, A. D. Schlüter and A. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6630–6640 CrossRef CAS.
- J. Xu, L. Tao, J. Liu, V. Bulmus and T. P. Davis, Macromolecules, 2009, 42, 6893–6901 CrossRef CAS.
- X. Hao, E. Malmström, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Aust. J. Chem., 2005, 58, 483–491 CrossRef CAS.
- A. Nyström and A. Hult, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3852–3867 CrossRef.
- A. Carlmark and E. Malmström, Macromolecules, 2004, 37, 7491–7496 CrossRef CAS.
- M. Malkoch, A. Carlmark, A. Woldegiorgis, A. Hult and E. Malmström, Macromolecules, 2004, 37, 322–329 CrossRef CAS.
- S. R. Benhabbour, M. C. Parrott, S. E. A. Gratton and A. Adronov, Macromolecules, 2007, 40, 5678–5688 CrossRef CAS.
- M. Rahm, R. Westlund, C. Eldsäter and E. Malmström, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6191–6200 CrossRef CAS.
- J. L. Hedrick, M. Trollsås, C. J. Hawker, B. Atthoff, H. Claesson, A. Heise and R. D. Miller, Macromolecules, 1998, 31, 8691–8705 CrossRef CAS.
- A. Nyström, M. Malkoch, I. Furó, D. Nyström, K. Unal, P. Antoni, G. Vamvounis, C. Hawker, K. Wooley, E. Malmström and A. Hult, Macromolecules, 2006, 39, 7241–7249 CrossRef.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- G. Moad and C. Barner-Kowollik, The Mechanism and Kinetics of the RAFT Process, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008 Search PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 301, 3661 CrossRef.
- P. Dubois, O. Coulembier and J.-M. Raquez, Handbook of Ring-Opening Polymerization, Wiley-VCH, Weinheim, 2009 Search PubMed.
- A.-C. Albertson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- J. Chiefari, G. Moad, E. Rizzardo, A. A. Gridnev, US Pat., 98/47927, 1998.
- J. Chiefari, J. Jeffery, R. T. A. Mayadunne, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 7700–7704 CrossRef CAS.
- A. N. Nikitin, R. A. Hutchinson, M. Buback and P. Hesse, Macromolecules, 2007, 40, 8631–8641 CrossRef CAS.
- T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7585–7605 CrossRef CAS.
- R. X. E. Willemse, A. M. van Herk, E. Panchenko, T. Junkers and M. Buback, Macromolecules, 2005, 38, 5098–5103 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., 2011, 50, 60–20 CAS.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Polymer, 2004, 45, 7791–7805 CrossRef CAS.
- T. Junkers, F. Bennet, S. P. S. Koo and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3433–3437 CrossRef CAS.
- A.-M. Zorn, T. Junkers and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 2028–2035 CrossRef CAS.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 201, 987–992 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1792–1798 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., 2009, 48, 2411–2414 CAS.
- C. Strazielle, H. Benoit and O. Vogl, Eur. Polym. J., 1978, 14, 331–334 CrossRef CAS.
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Anal. Chem., 2008, 80, 6915–6927 CrossRef CAS.
- P. Wu, M. Malkoch, J. Hunt, R. Vestberg, E. Kaltgrad, M. G. Finn, V. V. Fokin, K. B. Sharpless and C. J. Hawker, Chem. Commun., 2005, 5775–5777 RSC.
- M. Malkoch, K. Schleicher, E. Drockenmuller, C. J. Hawker, T. P. Russell, P. Wu and V. V. Fokin, Macromolecules, 2005, 38, 3663 CrossRef CAS.
- J. P. Van Hook and A. V. Tobolsky, J. Am. Chem. Soc., 1958, 80, 779 CrossRef CAS.
- S. P. S. Koo, T. Junkers and C. Barner-Kowollik, Macromolecules, 2009, 42, 62–69 CrossRef CAS.
- T. Junkers, Aust. J. Chem., 2008, 61, 646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further analytical data ((SEC-)ESI-MS and SEC as well as 1H-NMRspectra) for the dendronized acrylates and macromonomers. See DOI: 10.1039/c0py00411a |
| This journal is © The Royal Society of Chemistry 2011 |