Synthesis of glycopolypeptides by the ring opening polymerization of O-glycosylated-α-amino acid N-carboxyanhydride (NCA)†
Debasis
Pati
a,
Ashif Y.
Shaikh
b,
Srinivas
Hotha
*bc and
Sayam Sen
Gupta
*a
aCReST, Chemical Engineering Division, National Chemical Laboratory, Pune, India. E-mail: ss.sengupta@ncl.res.in; Fax: +91 20 2590 2021; Tel: +91 20 2590 2747
bDivision of Organic Chemistry, National Chemical Laboratory, Pune, India. E-mail: ay.shaikh@ncl.res.in; Tel: +91 20 2590 2331
cDepartment of Chemistry, Indian Institute of Science Education & ResearchE-mail: s.hotha@iiserpune.ac.in
First published on 16th February 2011
Abstract
The novel synthesis of O-glycosylated lysine-NCA from a stable glycosyl donor and a commercially available protected amino acid in very high yield is reported. These O-glycosylated lysine-NCA monomers underwent ring opening polymerization using simple primary amine initiators to form well defined, high molecular weight homoglycopolypeptides and diblock co-glycopolypeptides. The synthesis of azide labelled end functionalized glycopolypeptides and amphiphilic diblock copolypeptides is also reported. This methodology represents an easy and practical route to the synthesis of O-glycosylated polypeptides with 100% glycosylation.
Carbohydrates play a key role in a myriad of biological processes including inflammation, cell–cell contacts, signal transmission, fertilization and protein folding. In particular saccharides that are conjugated to proteins, commonly known as glycoproteins, are functionally very important in biology and there have been a lot of efforts directed for their efficient synthesis.1 Since biological synthesis of such glycoproteins is still very complex, artificial glycoconjugates provide an interesting biomimetic analogue. In this regard glycopolymers, featuring synthetic macromolecules with pendant carbohydrate moieties, have found widespread application in various fields such as macromolecular drug delivery systems, hydrogels, matrices for controlled cell culture, and as models of biological systems.2 Majority of these glycopolymers are acrylate based and controlled radical polymerization is used to synthesize polymers with controlled molecular weight, glycosylation density, and position—attributes that are necessary for biological recognition processes. However, these polymers do not have well-defined higher order structures and often adopt a random-coil conformation which inevitably renders some of the side-chain bioactive moieties inaccessible toward biological active sites. On the other hand, glycopolypeptides (glycopolymers with pendant carbohydrates on a polypeptide backbone) not only have the ability to fold into well-defined secondary structures3 (e.g., helix) but also mimic the molecular composition of proteoglycans. Therefore it is desirable to develop methodologies that afford easy and well defined synthetic glycopolypeptides.
Although well defined polypeptides based on natural and unnatural amino acids have been very successfully synthesized by the ring opening polymerization of their corresponding N-carboxyanhydrides (NCAs),4 the synthesis of glycopolypeptide still remains a major challenge. Glycopolypeptides can be synthesized by (1) ring opening polymerizations of glycosylated N-carboxyanhydride (NCA) monomers or (2) post-polymerization modification of polypeptide side chains. The first synthesis of glycopolypeptides using the ring opening polymerization of O-linked glycoserine NCA was reported quite some time back by Okada et al.5 However, the synthesis of the NCA monomer was very inefficient and required the usage of toxic Hg salts for the key glycosylation step.6 Further, the polymerization was extremely slow and limited to low degrees of polymerization. This was attributed to the steric effects and H-bonding between the sugar residue and NCA ring.5 The synthesis of the monomer has been improved upon by Cameron et al., but no polymerization of the synthesized O-linked glycoserine NCA was reported subsequently.7 Very recently, Deming and Kramer have synthesized C-linked glycosylated-L-lysine NCA monomers and have successfully polymerized it using transition metal initiators.8 Synthesis of glycopolypeptides by post-polymerization modification of synthetic polypeptides on the contrary has been more successful and several methods have been reported recently. These methods include coupling of β-D-galactosylamine to the carboxylic acid group of poly-L-glutamic acid9 to more recent reports of azide–alkyne cycloaddition reaction10 and thiol–ene click reactions11 for synthesis of glycopolypeptides. Although glycopeptides have been successfully synthesized by these methodologies, their main drawback is the incomplete sugar functionalization which is more predominant for high molecular weight polypeptides. Hence there is a need to develop simple and efficient methodologies to synthesize glycosylated amino acid NCA's which can be then polymerized to afford high molecular weight glycopolypeptides.
In this paper we report a novel and simple methodology for the synthesis of O-glycosylated-L-lysine NCA and their subsequent polymerization to afford glycopolypeptides. The O-glycosylated-L-lysine NCA that was synthesized has several attributes. First, the presence of the lysine side chain would put the sugar residue much farther away from the NCA ring. This would in turn reduce the sterics that were responsible for the inefficient polymerization of O-linked glycoserine NCA. Further, ROP of lysine NCA bearing various protecting groups has been extensively investigated and shown to undergo very efficient ROP to yield very high molecular weight polypeptides. Finally, the O-glycoside linkage between the sugar and the lysine side chain resembles more closely to the native linkages. The O-glycosylated-L-lysine NCA synthesized by our methodology underwent ring opening polymerization into well defined high molecular weight glycopolypeptide and diblock glycopolypeptide copolymers using simple amine initiators.
O-Linked glycopolypeptides synthesized by the NCA polymerization have not received the same attention as glycopolymers synthesized from acrylates and methacrylates since the critical glycosidation step for the synthesis of amino acid carbohydrate conjugate is very inefficient. The key to the successful synthesis of glycopolypeptides lies in development of a methodology that allows synthesis of glycosylated amino acids in very high yield. From our laboratories, we identified propargyl glycosides as novel glycosyl donors for transglycosylations using AuX3 (X = Cl or Br) as the catalyst in a non-diastereoselective fashion.12 Subsequently, the method was modified to give 1,2-transglycosides only by the use of propargyl 1,2-orthoesters in the presence of AuBr3/4 Å molecular sieves powder at room temperature.
In continuation of this programme on the use of gold catalysts for glycosylations, we observed unusual behaviour of Boc-protected amines with catalytic HAuCl4 while preparing serine-glycosides in a separate endeavour. t-Boc protected serine derivatives gave glycosyl carbamate instead of required O-glycoside in the presence of AuBr3/CH2Cl2/4 Å MS powder/rt and further experiments gave us optimized conditions for getting glycosyl carbamates as exclusive products with HAuCl4 as the catalyst in CH2Cl2 at room temperature.13 Normally the tert-butyl group of the Boc protecting group would fall off in the presence of acids and liberate CO2 to give the amine. Contrary to this, Au(III) catalyzed glycosylation reaction resulted in the formation of a glycosyl carbamate. Thus, we envisioned that ε-Boc protected CbzLysOBn would be ideal to prepare amino acid glycoconjugate which can subsequently be converted to the corresponding glycosyl carbamate using gold catalyzed glycosylation procedure. Accordingly, glycosylation reaction between ε-Boc protected CbzLysOBn and propargyl 1,2-orthoester of glucose (1a) and mannose (1b) was conducted in the presence of HAuCl4 and 4 Å molecular sieves powder in CH2Cl2 at room temperature to afford the carbamates 2a and 2b in 95% and 92% yield respectively (Scheme 1). Furthermore, we continued our journey towards the glyco-NCA in two steps: first, we subjected the glycoconjugates 2a and 2b to hydrogenation using 10% Pd/C at 400 psi to obtain per-O-benzoylated-D-glucose-L-lysine carbamate or per-O-benzoylated-D-mannose-L-lysine carbamate. They were then subsequently converted to their corresponding NCA's 3a (β-gluco-O-Lys) and 3b (α-manno-O-Lys) using triphosgene and α-pinene7 in 80% yield after three crystallizations (Scheme 1). The purified NCA's 3a and 3b were thoroughly characterized by NMR spectroscopic studies. As delineated above, the major hurdle for the synthesis of biomimetic glycopolypeptides has been the limited access to this important class of glyco amino acid NCA's. Literature methods suffer from one or more of (i) toxic metals for the activation, (ii) long reaction times, (iii) laborious purification procedures and (iv) poor yields. A recent report revealed glycopolypeptides with lysine backbone from a known C-glycosyl precursor in a multistep manner with a poor overall yield of 25%.8 On the other hand, our current endeavour for the synthesis of glyco amino acid NCA is novel, has near quantitative yield, uses catalytic quantity of HAuCl4 and involves a very simple purification procedure.
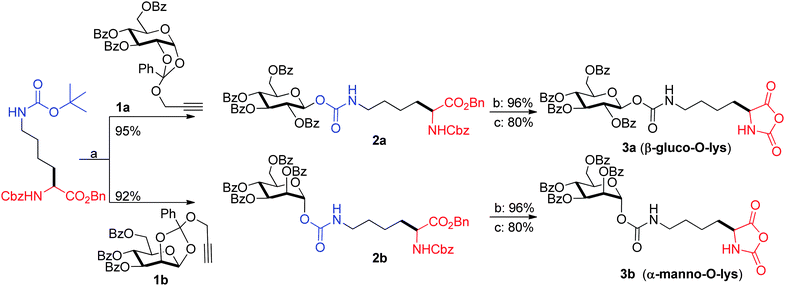 | ||
| Scheme 1 Synthesis of glyco amino acid NCA's. (a) HAuCl4, CH2Cl2, rt, 0.5 h, 4 Å MS powder; (b) Pd/C, H2, 400 psi, CH3OH, 12 h; (c) triphosgene, THF, α-pinene, 70 °C. | ||
Polymerization of 3a (β-gluco-O-Lys NCA) was first attempted using hexylamine as the initiator (M/I = 25) in dry acetonitrile (Scheme 2). The progress of the polymerization was followed by monitoring the disappearance of the anhydride stretch of the NCA ring at 1787 and 1852 cm−1.
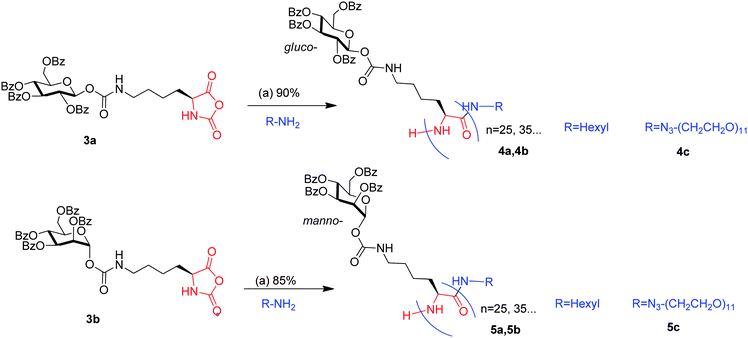 | ||
| Scheme 2 Glycopolypeptides by ROP of glyco amino acid NCA's. (a) Dioxane/acetonitrile/DMF, proton sponge (0.25 eq.), 24 h, rt. | ||
However, no decrease in the anhydride stretch was observed even after 48 h of initiation. Similar observation was noted even when the solvent was changed to dry dioxane or DMF. We reasoned that there might be very small amounts of residual acid left which protonated the primary amine initiator to inhibit the initiation of the polymerization reaction. To remove the residual acid, we proceeded with the polymerization again in the presence of a small amount of non-nucleophilic base 1,8-bis(dimethylamino)naphthalene “proton sponge” (0.25 eq. with respect to monomer; run 1). The FT-IR of this reaction mixture after 24 h showed complete disappearance of the NCA anhydride stretch indicating that the polymerization reaction proceeded to completion. However, after 6 h, the reaction mixture which was homogeneous during the onset of the polymerization became heterogeneous and white powdery precipitate was observed. The number average molecular weight of the precipitated polymer 4a was estimated to be 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by GPC and a broad molecular weight distribution with a PDI of 1.5 was observed (Table 1, run 1). Control reactions in which 3a was incubated with only 1,8-bis(dimethylamino)naphthalene (0.25 to 1 eq. of 3a) showed no change in the anhydride IR stretch even after 48 h thereby showing that the primary amine initiator was only responsible for initiation of the polymerization reaction. Since 1,8-bis(dimethylamino)naphthalene did not initiate polymerization, we used this as an additive in all the subsequent polymerization reactions.
000 by GPC and a broad molecular weight distribution with a PDI of 1.5 was observed (Table 1, run 1). Control reactions in which 3a was incubated with only 1,8-bis(dimethylamino)naphthalene (0.25 to 1 eq. of 3a) showed no change in the anhydride IR stretch even after 48 h thereby showing that the primary amine initiator was only responsible for initiation of the polymerization reaction. Since 1,8-bis(dimethylamino)naphthalene did not initiate polymerization, we used this as an additive in all the subsequent polymerization reactions.
| Run no. | Monomer (M) | Initiator | Solvent | Product polymer | |||||
|---|---|---|---|---|---|---|---|---|---|
| Initiator (I) | M/Ia | Polymer | M n b | M w/Mnb | DP c | Yieldd | |||
| a M/I indicates monomer to initiator ratio. b Molecular weight and polydispersity index were estimated from GPC. c Degree of polymerization (DP) from GPC. d Total isolated yield. | |||||||||
| 1 | β-gluco-O-Lys | Hexylamine | 25 | CH3CN | 4a | 73 000 | 1.50 | 97 | 90% |
| 2 | β-gluco-O-Lys | Hexylamine | 25 | Dioxane | 4a | 31 300 | 1.06 | 41 | 95% |
| 3 | β-gluco-O-Lys | Hexylamine | 35 | Dioxane | 4b | 34 900 | 1.12 | 46 | 95% |
| 4 | α-manno-O-Lys | Hexylamine | 25 | Dioxane | 5a | 52 000 | 1.09 | 69 | 95% |
| 5 | α-manno-O-Lys | Hexylamine | 35 | Dioxane | 5b | 67 400 | 1.10 | 89 | 95% |
| 6 | α-manno-O-Lys | Hexylamine | 25 | DMF | 5a | 34 800 | 1.08 | 46 | 95% |
| 7 | α-manno-O-Lys | Hexylamine | 35 | DMF | 5b | 38 500 | 1.12 | 51 | 95% |
| 8 | β-gluco-O-Lys | N3PEGNH2 | 25 | DMF | 4c | 34 000 | 1.11 | 45 | 90% |
| 9 | α-manno-O-Lys | N3PEGNH2 | 25 | DMF | 5c | 16 900 | 1.22 | 22 | 80% |
Since a broad molecular weight distribution of 4a was observed and the molecular weight obtained was much higher than expected, the polymerization of 3a was attempted again in dry dioxane using hexylamine as the initiator (M/I = 25, 35) in the presence of 0.25 eq. of 1,8-bis(dimethylamino)naphthalene “proton sponge” (Table 1, runs 2 and 3). Both the polymerization reactions went to completion in 24 h as was observed by FT-IR. The resulting polymers 4a and 4b were purified by reprecipitation and the structures were identified by 1H and 13C NMR (ESI†). The molecular weight distribution observed from GPC was monomodal and found to be reasonably narrow (Fig. 1). The Mn was estimated to be 31300 and 34900 while the PDI was calculated to be 1.06 and 1.12 for 4a and 4b respectively. The higher molecular weight that is observed is due to incomplete initiation by hexylamine as has been observed before. We then attempted the polymerization of 3b (α-manno-O-Lys NCA) with hexylamine as the initiator (M/I = 25, 35) in dioxane (Scheme 2; Table 1, runs 4 and 5). The molecular weight distribution was again found to be reasonably narrow although the molecular weights of the resulting polymers 5a and 5b were estimated to 52000 and 67400 respectively. The polymers were also thoroughly characterized by 1H and 13C NMR (ESI†). To study the effect of polar solvent on the polymerization reaction, we carried out the same polymerization in dry DMF (Table 1, runs 6 and 7). The resulting polymers formed in DMF had a molecular weight which was much closer to the expected molecular weights based on the M/I ratio. This is in contrast to the recently reported polymerization of C-glycosyl NCA's where considerably higher molecular weights (approximately 3 times) were reported for the glycopolypeptides synthesized using organometallic Co initiators. To prove that the initiator was incorporated into the polymer, we carried out the polymerization of 3a and 3b in DMF with azide–PEG–NH2 (n = 11) as the initiator. The resultant polymers 4c and 5c were purified by multiple reprecipitation and then characterized by NMR and FT-IR. The FT-IR of 4c and 5c show sharp peak at 2110 cm−1 that is characteristic for the organo azide stretch (S3, ESI†). Usage of this bifunctional azide–PEG–amine initiator allows the synthesis of end functionalized polymer which can be further manipulated using Cu(I) catalyzed azide–alkyne “click chemistry”.14
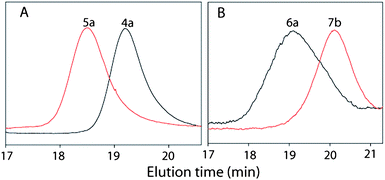 | ||
| Fig. 1 Size exclusion chromatogram of (A) homopolypeptides 4a and 5a synthesized in dioxane and (B) diblock copolypeptides 6a and 7b (DMF/0.1 M LiBr, 60 °C, RI). | ||
Diblock copolymers were prepared by combining 3a and 3b with conventional NCA's as shown in Scheme 3. For example, the polymerization of p-methoxybenzyl-L-glutamate (PMBn–glu) NCA (3e) was first initiated with hexylamine as initiator (M/I = 15) in DMF. After completion of the first stage polymerization (3 h) as observed by FT-IR, the second monomer 3a (M1:M2 = 15:25) and “proton sponge” were added to the reaction mixture (Table 2, run 1). The polymerization proceeded to completion in high conversion to yield polymer 6a in predictable monomer composition as observed by 1H NMR (S1, ESI†). The ratio of PMBn–glu and β-gluco-O-Lys was estimated to be 1:1.5 (expected 1:1.7) from their characteristic signals in the 1H NMR. The GPC showed narrow molecular weight distribution with a PDI of 1.15. The polymer 6a was then treated with 10% TFA in dichloromethane at rt for 30 min to deprotect the p-methoxybenzyl group. The polymer 9 thus formed was then characterized by 1H and 13C NMR to confirm the complete deprotection of the p-methoxybenzyl group (ESI†). Thus we were able to synthesize an amphiphilic diblock co-polymer having a hydrophobic benzoate protected glucose and a hydrophilic carboxylic acid on the polypeptide backbone. Similarly, other diblock copolypeptides were synthesized with 3b and conventional NCA's like γ-benzylglutamate NCA and Z-Lys NCA in high yield with narrow molecular weight distribution (Table 2). The presence of the initiator in the block copolymer was again confirmed by synthesizing a block co-polymer of 3b and γ-benzylglutamate NCA with azide–PEG–NH2 (n = 11) as the initiator (Table 2, run 4). The presence of the 2100 cm−1 organo azide stretch in FT-IR for the resultant polymer indicated that the initiator was successfully incorporated into the polymer chain. The amount of azide–PEG–NH2 incorporated into this block copolymer was estimated by attaching fluorescein alkyne to the azide functionalized polymer 7b using Cu(I) catalyzed azide–alkyne “click chemistry” (ESI†). The amount of azide groups incorporated was estimated from UV-VIS spectroscopy to be around 0.75 mol mol−1 of polymer 7b. Hence, we were also able to synthesize fluorescently labelled block co-polypeptides using this methodology.
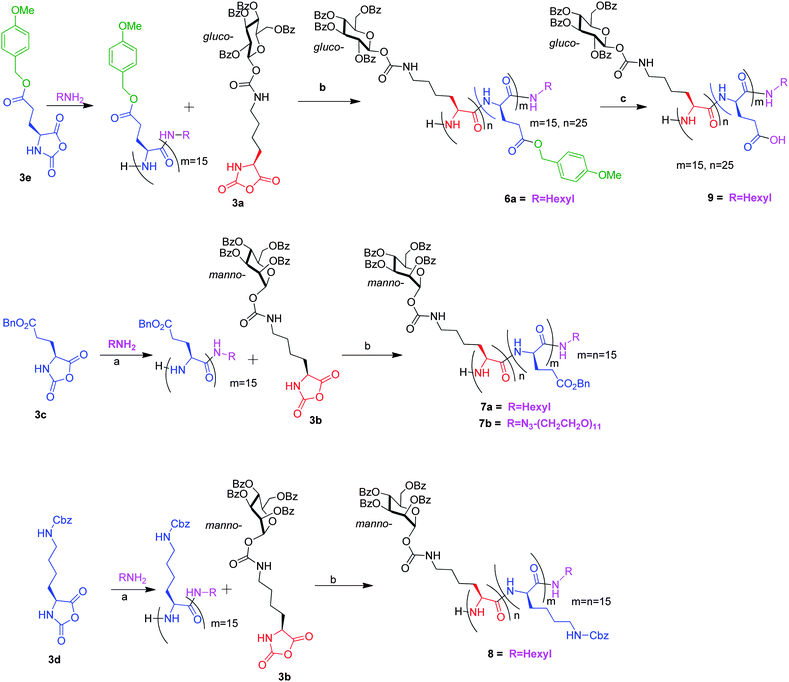 | ||
| Scheme 3 Synthesis of diblock copolypeptides. (a) DMF, rt; (b) DMF, 0.25 eq. Proton Sponge, 30 h, rt, 95%; (c) 10%TFA in CH2Cl2, 0.5 h. | ||
| Run no. | First monomer (M1) | Initiator | Product polymer | |||||
|---|---|---|---|---|---|---|---|---|
| Second monomer (M2) | [M1]:[M2]/I |
Polymer | Expected Mn × 103 | Observed Mn × 103b | M w/Mnb | Yieldc | ||
| a For run 4, azide–PEG–NH2 (n = 11) was used as the initiator. b Molecular weight and polydispersity index were estimated from GPC. c Total isolated yield. | ||||||||
| 1 | PMBn-glu | β-gluco-O-Lys | 15:25 |
6a | 22.895 | 36.0 | 1.15 | 90% |
| 2 | Bn-L-glu | α-manno-O-Lys | 15:15 |
7a | 14.695 | 16.5 | 1.11 | 95% |
| 3 | Z-Lys | α-manno-O-Lys | 15:15 |
8 | 15.295 | 16.4 | 1.14 | 95% |
| 4a | Bn-L-glu | α-manno-O-Lys | 15:15 |
7b | 15.165 | 19.0 | 1.07 | 95% |
Conclusions
We have reported a very easy three step synthesis of an O-glycosylated lysine-NCA using a stable glycosyl donor and a commercially available protected amino acid. The highlight of the synthesis is that the key glycosylation step and the subsequent deprotection reaction proceed to completion in near quantitative yield. This ease of synthesis allows us to synthesize these monomers in very high yield and we believe would be useful to several groups interested in synthesis of glycopeptides. The glycosylated NCA's were then polymerized using commercially available simple amine initiators to yield well defined high molecular weight homopolypeptides and diblock copolypeptides in very high yields. Addition of 1,8-bis(dimethylamino)naphthalene (“proton sponge”) was necessary for the successful completion of the polymerization reaction, although its exact role is not yet fully understood. We were also able to synthesize end-functionalized, amphiphilic and fluorescently labelled polymers using our methodology. Saponification of the esters from the sugar residues of the synthesized glycopolypeptides and further lectin binding studies on the resulting water soluble glycopolypeptides are currently underway in our laboratories. Results from those studies would be communicated in due course of time.Experimental section
Materials and methods
Propargyl 1,2-orthoesters 1a and 1b were prepared according to the literature procedure.12bCbzLys(Boc)OH was obtained from Aldrich and converted to CbzLys(Boc)OBn using standard literature procedure. HAuCl4, p-methoxy benzyl alcohol, hexylamine, triphosgene and azide–PEG–amine (n = 11) were obtained from Aldrich. All other chemicals used were obtained from Merck, India. Diethyl ether, petroleum ether (60–80 °C), ethylacetate, dichloromethane, tetrahydrofuran, and dioxane were brought from Merck and dried by conventional methods and stored in the glove box. DMF (99.99% dry) obtained from Sigma Aldrich was used for polymerization inside the glove box. FT-IR spectra were recorded on a Perkin Elmer FT-IR spectrum GX instrument by making KBr pellets. Pellets were prepared by mixing 3 mg of sample with 97 mg of KBr. 1H NMR spectra were recorded on Bruker Spectrometers (200 MHz, 400 MHz or 500 MHz). 13C NMR and DEPT spectra were recorded on a Bruker Spectrometer (50 MHz, 100 MHz or 125 MHz) and reported relative signals according to the deuterated solvent used. Size-exclusion chromatography of the glycopolypeptides was performed using an instrument equipped with a Waters 590 pump with a Spectra System RI-150 RI detector. Separations were effected by 105 and 103 Å Phenomenex 5 µ columns using 0.1 M LiBr in DMF eluent at 60 °C at a sample concentration of 5 mg ml−1. A constant flow rate of 1 ml min−1 was maintained, and the instrument was calibrated using polystyrene standards. UV-VIS spectra were obtained.General procedure for the synthesis of amino acid glycosyl carbamates (2a and 2b)
To a solution of propargyl 1,2-orthoester12b (1a or 1b, 0.1 mmol), CbzLys(Boc)OBn (0.11 mmol) and activated 4 Å molecular sieves powder (50 mg) in anhydrous CH2Cl2 (5 ml) was added HAuCl4 (7 mol%) under argon atmosphere at room temperature. The reaction mixture was stirred at room temperature for the specified time and the reaction mixture was filtered and the filtrate was concentrated in vacuo. The resulting residue was purified by silica gel column chromatography using ethyl acetate/petroleum ether as the mobile phase to afford the compounds 2a and 2b.General procedure for the synthesis of glyco N-carboxyanhydrides (3a and 3b)
Hydrogenolysis of compounds 2a and 2b was carried out using 10% Pd/C in MeOH/EtOAc (9:1) at 400 psi for 12 h. After completion of the reaction, the reaction mixture was filtered and concentrated under reduced pressure to afford per-O-benzoylated-D-glucose-L-lysine carbamate or per-O-benzoylated-D-mannose-L-lysine carbamate in almost quantitative yield. The resulting compounds were directly used for NCA synthesis without any further purification.
To a solution of per-O-benzoylated-D-glucose-L-lysine carbamate or per-O-benzoylated-D-mannose-L-lysine carbamate (654 mg, 0.85 mmol) in freshly distilled out tetrahydrofuran (10 ml) was added a solution of triphosgene (126 mg, 0.425 mmol) in anhydrous tetrahydrofuran (2 ml) under argon and the reaction mixture was heated to 50–55 °C. α-Pinene (0.202 ml, 1.28 mmol) was then added and the reaction mixture was allowed to stir for an additional 2 h. The reaction mixture was then cooled to room temperature and then poured into dry hexane (300 ml) to afford a white precipitate, which was filtered off quickly and crystallized two more times using a mixture of ethyl acetate and petroleum ether. Finally, the white precipitate of glyco N-carboxyanhydride (3a or 3b) was dried under vacuum and transferred into the glove box. Final yield 544 mg (80%).
General procedure for the synthesis of glycopolypeptides
To a solution of glyco-L-lysine NCA 3a or 3b (100 mg ml−1) in dry dioxane, acetonitrile or DMF was added with “proton sponge” 1,8-bis(dimethylamino)naphthalene (0.25 equivalent to monomer, 1 M) as an additive and hexylamine or azide–PEG–amine (0.5 M) as the initiator inside the glove box. The progress of the polymerization was monitored by FT-IR spectroscopy by comparing with the intensity of the initial NCA's anhydride stretching at 1789 cm−1 and 1852 cm−1. The reactions generally completed within 24 to 30 h. Aliquots were removed after completion of polymerization for GPC analysis. Finally the solvent was removed under reduced pressure from the reaction mixture. The resulting residue was redissolved in DCM and then the polymer precipitated out by addition of methanol. The precipitated polymer was collected by centrifugation and dried to afford white glycopolypeptides 4a, 4b, 4c, 5a, 5b and 5c in almost 85–90% yield.General procedure for the synthesis of diblock coglycopolypeptides
To a solution of the first NCA monomer (15 eq., 100 mg ml−1) in DMF was added hexylamine (1 eq.) at rt inside the glove box. The progress of the reaction was monitored by FT-IR. Upon near consumption of the first monomer (>95%), the second glyco amino acid NCA (25 eq. of 3a or 15 eq. of 3b) together with the proton sponge (0.25 eq. of the second NCA monomer) in DMF (100 mg ml−1) was added and the progress of the reaction was monitored by FT-IR. The reactions generally completed within 30 h. Aliquots were removed after completion of polymerization for GPC analysis. Finally the solvent was removed under reduced pressure from the reaction mixture. The resulting residue was redissolved in DCM and then the polymer precipitated out by addition of methanol. The precipitated polymer was collected by centrifugation and dried to afford white diblock glycopolypeptides 6a, 7a, 7b and 8 in almost 85–90% yield.Acknowledgements
The authors acknowledge Dr P Rajmohanan for NMR support and Mrs D. Dhoble for help with GPC. DP and ASY thank CSIR, New Delhi for research fellowship. SSG thanks CSIR Network project NWP0051-C and SH thanks DST, New Delhi for SwarnaJayanti Fellowship.Notes and references
- (a) D. B. Werz and P. H. Seeberger, Nature, 2007, 446, 1046–1051 CrossRef CAS; (b) D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS.
- (a) L. Albertin and N. R. Cameron, Macromolecules, 2007, 40, 6082–6093 CrossRef CAS; (b) G. Chen, L. Tao, G. Mantovani, J. Geng, D. Nyström and D. M. Haddleton, Macromolecules, 2007, 40, 7513–7520 CrossRef CAS; (c) V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS; (d) V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS; (e) S. G. Spain, M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059–2072 CrossRef CAS; (f) S. R. S. Ting, E. H. Min, P. Escalé, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422–9434 CrossRef CAS.
- T. J. Deming, Soft Matter, 2005, 1, 28–35 RSC.
- (a) N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS; (b) T. J. Deming, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3011–3018 CrossRef CAS.
- (a) K. Aoi, K. Tsutsumiuchi and M. Okada, Macromolecules, 1994, 27, 875–877 CrossRef CAS; (b) K. Aoi, K. Itoh and M. Okada, Macromolecules, 1995, 28, 5391–5393 CrossRef CAS; (c) K. Aoi, K. Tsutsumiuchi, E. Aoki and M. Okada, Macromolecules, 1996, 29, 4456–4458 CrossRef CAS; (d) K. Tsutsumiuchi, K. Aoi and M. Okada, Macromolecules, 1997, 30, 4013–4017 CrossRef CAS.
- E. Rüde, O. Westphal, E. Hurwitz, S. Fuchs and M. Sela, Immunochemistry, 1966, 3, 137–151 CrossRef CAS.
- M. I. Gibson, G. J. Hunt and N. R. Cameron, Org. Biomol. Chem., 2007, 5, 2756–2757 RSC.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2010, 132, 15068–15071 CrossRef CAS.
- Y. Wang and K. L. Kiick, J. Am. Chem. Soc., 2005, 127, 16392–16393 CrossRef CAS.
- J. Huang, G. Habraken, F. Audouin and A. Heise, Macromolecules, 2010, 43, 6050–6057 CrossRef CAS.
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445–4448 CrossRef CAS.
- (a) S. Hotha and S. Kashyap, J. Am. Chem. Soc., 2006, 128, 9620–9621 CrossRef CAS; (b) G. Sureshkumar and S. Hotha, Tetrahedron Lett., 2007, 48, 6564–6568 CrossRef CAS; (c) G. Sureshkumar and S. Hotha, Chem. Commun., 2008, 4282–4284 RSC.
- A. Y. Shaikh, G. Sureshkumar, D. Pati, S. S. Gupta, S. Hotha, 2010, submitted.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectral charts of all newly synthesized compounds. See DOI: 10.1039/c0py00412j |
| This journal is © The Royal Society of Chemistry 2011 |