DOI:
10.1039/C0PY00395F
(Paper)
Polym. Chem., 2011,
2, 1146-1155
Star and miktoarm star block (co)polymers viaself-assembly of ATRP generated polymer segments featuring Hamilton wedge and cyanuric acid binding motifs†
Received
3rd December 2010
, Accepted 12th January 2011
First published on 16th February 2011
Abstract
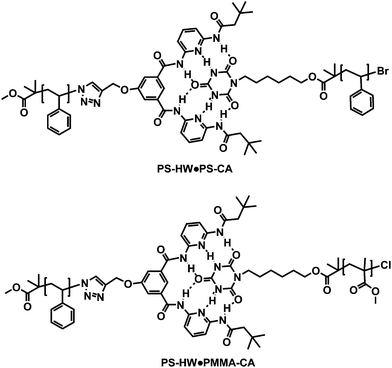
Hamilton wedge (HW) end-functionalized poly(styrene) (PS–HW, Mn = 5400 g mol−1, PDI = 1.06), HW mid-chain functionalized poly(styrene) (PS–HW–PS, Mn = 4600 g mol−1, PDI = 1.04), cyanuric acid (CA) end-functionalized poly(styrene) (PS–CA, Mn = 3700 g mol−1, PDI = 1.04) and CA end-functionalized poly(methyl methacrylate) (PMMA–CA, Mn = 8500 g mol−1, PDI = 1.13) precursors were successfully synthesized via a combination of atom transfer radical polymerization (ATRP) and copper catalyzed azide–alkynecycloaddition (CuAAC). The precursor polymers were characterized viasize exclusion chromatography (SEC) and 1H NMR with respect to both molecular weight and structure. Supramolecular homopolymer (PS–HW·PS–CA), block copolymer (PS–HW·PMMA–CA), star polymer (PS–HW–PS·PS–CA) as well as miktoarm star polymer (PS–HW–PS·PMMA–CA) were formed in solution in high yields at ambient temperature (association close to 89% for PS–HW·PS–CA, 90% for PS–HW–PS·PS–CA and 98% for PS–HW–PS·PMMA–CA) via H-bonding between the orthogonal recognition units, HW and CA. The formation of supramolecular polymers was confirmed via1H NMR at ambient temperature in deuterated methylene chloride (CD2Cl2) solution.
Introduction
A branched polymer structure is described as a non-linear polymer with multiple backbone chains growing from junction points.1,2 It has been demonstrated that branching results in a more compact structure in comparison to linear counterparts of similar molecular weight, due to the higher segment density of the branched structure, which changes the melt, solution and solid-state properties of the polymer.1 Star polymers have been generally prepared by living ionic polymerization techniques until recently.3,4 In the last decade, with an advance in the living radical polymerization (LRP) routes,5 the facile synthesis of both linear and star polymers became possible and received widespread attention due to the variety of applicable monomers and greater tolerance to experimental conditions in comparison with living ionic polymerization routes.6,7 In recent years, orthogonal conjugation reactions and their combination with LRP techniques have been increasingly applied towards the preparation of both linear and star polymers due to their efficiency, orthogonality to various functional groups, simple workup as well as compatibility with LRP.8–11 The connectivity between the individual structural elements of the complex architecture structure is thus covalent. Supramolecular chemistry—in contrast—is characterized by non-covalent interactions, which play a key role in the assembly, conformation, and/or behavior of supramolecular systems.12 These reversible non-covalent interactions mainly include H-bonding,13 metal–ligand coordination,14 π–π stacking15 and ion–dipole interactions.16 In a similar way, supramolecular polymers can be classified as polymeric systems that utilize these reversible non-covalent interactions to define their assembly, conformation, and/or behavior.17–19 Thus, supramolecular polymers can be generated by a single or a combination of these non-covalent interactions described above. H-Bonding has proved to be one of the most prominent supramolecular motifs, due to its ease of accessibility and high binding constants.19Hydrogen bonds display sensitivity to temperature changes and can form stable associations in a range of solvents.19 H-Bonding or its combination with metal–ligand motif has been utilized for the generation of a wide variety of supramolecular polymers, such as homo, block copolymers (di- and multiblock), star, cyclic and dendrimeric polymers.20 However, relatively few studies focusing on the synthesis of supramolecular star polymersvia H-bonding interaction have been reported until now. Recently, Zimmerman and Todd produced linear poly(styrene) (PS) and polylactide terminated with a ditopic H-bonding module and converted these low molecular weight linear polymers into high molecular weight supramolecular star structures through a self-assembly process.21 More recently, Tam and co-workers reported the synthesis of linear poly(methyl methacrylate) (PMMA) with a guanosine terminal group generated via atom transfer radical polymerization (ATRP), followed by its self-assembly into the final supramolecular star polymer with eight arms via an addition of potassium picrate as a self-assembling partner.22a Tam and colleagues demonstrated that the star polymers obtained via divergent and convergent approaches were identical. Meanwhile, Bernard and colleagues reported the synthesis of supramolecular poly(vinyl acetate) (PVA), which was achieved via reversible addition–fragmentation chain transfer (RAFT) polymerized chains with H-bonding motifs—specifically thymine and diaminopyridine derivatives—located at the polymer chain's terminus as well as in a mid-chain position, respectively.23 The self-assembly of the PVA chains into an A3 star polymer in CDCl3 was successfully demonstrated by 1H NMR spectroscopy. The same group published the synthesis of AB2 type miktoarm star polymers using similar supramolecular motifs.20m Recently, we reported for the first time that α,ω-hydrogen donor/acceptor (HW/CA) functional polymer strands prepared via a combination of living radical polymerization (ATRP) and orthogonal conjugation are able to undergo self-assembly as single chains to emulate—on a simple level—the self-folding behavior of natural biomacromolecules.24
The characterization of supramolecular polymers19 is not feasible with most common polymer characterization techniques due to the fact that H-bonding depends on the solvent, concentration, temperature and pressure. However, 1H NMR is an efficient, non-invasive and well-known technique to characterize the H-bonding directed self-assembly of polymers, allowing the study of a wide range of concentrations and temperatures.18
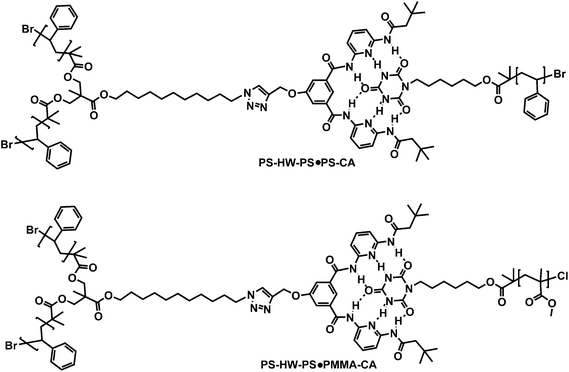
To the best of our knowledge, there are no previously reported examples of star or miktoarm star polymers prepared using highly binding recognition (HW/CA) units through the combination of ATRP and click chemistry.25 These six-hydrogen bonding motifs were deliberately chosen, as they have extremely high association constants (Kass = 106 M−1 in CHCl3).20e When these systems are compared with other motifs employed to produce supramolecular stars (such as the three-hydrogen bonding thymine and diaminopyridine motifs (Kass = 120–140 M−1 in CDCl3)20m employed by Bernard and colleagues) there is an over four order of magnitude increase in the observed association constants. Moreover, the results reported herein provide—for the first time—a calculation of the degree of formation of these supramolecular assembliesvia1H NMR spectroscopy. In the present contribution, we report the synthesis of well-defined Hamilton wedge (HW)26 mid- and end-functionalized poly(styrene)s (PS–HW–PS and PS–HW), as well as cyanuric acid end-functionalized poly(methyl methacrylate) (PMMA–CA) and poly(styrene) (PS–CA). These base polymers are carefully characterized viaNMR and size exclusion chromatography. Subsequently, the base macromolecular structures are self-assembled into supramolecular architectures, i.e.PS–HW–PS·PMMA–CA miktoarm star polymer, PS–HW–PS·PS–CA star polymer, PS–HW·PMMA–CAdiblock copolymer and PS–HW·PS–CA extended homopolymer in CD2Cl2 at ambient temperature, respectively. The self-assembly into the respective PS–HW–PS·PMMA–CA, PS–HW–PS·PS–CA, PS–HW·PMMA–CA and PS–HW·PS–CA structures was established via an extensive 1H NMR study. The targeted structures are depicted in Scheme 1 (block copolymers) and Scheme 2 (star and miktoarm star polymers). The molecular weight selection for the poly(styrene) and poly(methyl methacrylate)polymers in the range between 3700 ≤ Mn/g mol−1 ≥ 9000 is guided by the following considerations: (i) The lower molecular weight material (<3000 g mol−1) PS contains a HW, which is not soluble in common organic solvents. Thus, 3000 g mol−1 defines the lower molecular weight barrier. (ii) For the self-assembly systems, it is desirable to employ as pure as possible macromolecular building blocks. Unfortunately, when higher molecular weights are targeted (>9k Da), radical–radical coupling reactions occur during the synthesis of high molecular weight poly(styrene) by ATRP (see Fig. S11 in the ESI†). Therefore, the molecular weights employed in the current study present an optimum between solubility and purity.
Experimental section
The materials section as well as a detailed description of the analytical instrumentation can be found in the ESI†.
Syntheses
2,2,5-Trimethyl-1,3-dioxane-5-carboxylic acid (1),276-(2,4,6-trioxo-1,3,5-triazinan-1-yl)hexyl 2-bromo-2-methylpropanoate (8),24 mono azide end-functionalized PS (PS–N3) (11)28 and the N1,N3-bis(6-(3,3-dimethylbutanamido)pyridin-2-yl)-5-(prop-2-ynyloxy)isophthalamide (12)24 were synthesized according to the literature procedures.
Synthesis of 11-bromoundecyl 2,2,5-trimethyl-1,3-dioxane-5-carboxylate (2).
11-Bromoundecan-1-ol (1.00 g, 3.98 mmol), compound 1 (1.04 g, 5.97 mmol) and DMAP (0.073 g, 0.60 mmol) were dissolved in dry DCM (20 mL). DCC (1.85 g, 8.95 mmol) was dissolved in 10 mL dry DCM and subsequently added to the solution. The reaction was carried out at ambient temperature overnight. Solids were filtered off, the filtrate was concentrated and the crude product was purified viacolumn chromatography on silica gel, eluting with ethyl acetate/n-hexane (1/10) to obtain compound 2 as a viscous liquid (1.18 g, yield: 73%). 1H NMR (400 MHz, CDCl3) δ (ppm) 4.21–4.10 (q, 2H, CH2CH2OCO), 3.66–3.61 (d, 4H, CCH2O), 3.43–3.38 (t, 2H, BrCH2), 1.9–1.79 (m, 2H, BrCH2CH2), 1.67–1.58 (m, 2H, OCH2CH2), 1.43 (s, 3H, CCH3), 1.39 (s, 3H, CCH3), 1.28 (br m, 14H, (CH2)7), 1.2 (s, 3H, CCH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 176.05, 98.04, 68.69, 66.04 65.23, 49.06, 33.07, 32.17, 29.14, 29.13, 29.17, 29.75, 28.73, 28.51, 26.43, 26.43, 15.32. ESI-MS (M + Na)+ C19H35BrO4 theoretical: 429.16, experimental: 429.12.
Synthesis of 11-bromoundecyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (3).
Compound 2 (1.00 g, 2.45 mmol) was dissolved in 15 mL of THF and 15 mL of 1 M HCl (aq.) was slowly added to the solution. The reaction mixture was stirred for 4 h. The solution was diluted with DCM and then extracted two times with water. The combined organic phases were dried over Na2SO4, filtered and evaporated. The crude product was purified viacolumn chromatography on silica gel, eluting with ethyl acetate/n-hexane (1/10) to remove the residual unreacted compound 2, eluting with ethyl acetate/n-hexane (1/1) to obtain compound 3 as a white solid (0.83 g, yield: 93%). 1H NMR (400 MHz, CDCl3) δ (ppm) 4.19–4.14 (t, 2H, CH2CH2OCO), 3.94–3.89 (d, 2H, CCH2O), 3.74–3.69 (d, 2H, CCH2O), 3.43–3.38 (t, 2H, BrCH2), 2.45 (br s, 2H, OH), 1.9–1.79 (m, 2H, BrCH2CH2), 1.67–1.58 (m, 2H, OCH2CH2), 1.28 (br m, 14H, (CH2)7), 1.2 (s, 3H, CCH3). 13C NMR (100 Hz, CDCl3) δ. 13C NMR (100 MHz, CDCl3) δ (ppm) 176.05, 68.55, 65.23, 49.08, 34.05, 33.05, 29.42, 29.41, 29.17, 29.13, 28.83, 28.53, 28.71, 25.84, 17.15. ESI-MS (M + Na)+C16H31BrO4 theoretical: 389.13, experimental: 389.08.
Synthesis of 11-azidoundecyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (4).
Compound 3 (0.80 g, 2.17 mmol) was dissolved in 15 mL of DMF and NaN3 (0.7 g, 10.85 mmol) was added to the solution. The reaction mixture was stirred at 80 °C for 24 h and subsequently cooled to ambient temperature. Distilled water was added to the solution and the product was extracted three times with diethyl ether. The combined organic phases were dried over Na2SO4, filtered and evaporated. Product 4 was isolated as a white solid (0.71 g, yield: 99%). 1H NMR (400 MHz, CDCl3) δ (ppm) 4.19–4.14 (t, 2H, CH2CH2OCO), 3.94–3.89 (d, 2H, CCH2O), 3.74–3.69 (d, 2H, CCH2O), 3.28–3.23 (t, 2H, N3CH2), 2.76 (br s, 2H, OH), 1.69–1.57 (m, 4H, OCH2CH2 and N3CH2CH2), 1.28 (br m, 14H, (CH2)7), 1.2 (s, 3H, CCH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 176.05, 68.55, 65.23, 51.49, 49.08, 29.42, 29.41, 29.17, 29.13, 28.83, 28.53, 28.71, 25.84, 17.15. ESI-MS (M + Na)+C16H31N3O4 theoretical: 352.22, experimental: 352.12.
Synthesis of azide-functionalized ATRPinitiator (5).
Compound 4 (0.7 g, 2.12 mmol) was dissolved in 20 mL of dry THF. Et3N (1.2 mL, 8.51 mmol) and DMAP (0.052 g, 0.42 mmol) were added to the solution and cooled to 0 °C. 2-Bromoisobutyryl bromide (1.05 mL, 8.51 mmol) was dissolved in 10 mL of dry THF and subsequently added dropwise to the reaction mixture within 30 min. After the addition, the reaction mixture was warmed to ambient temperature and stirred further for 5 h. Solids were filtered off, the filtrate was concentrated and diluted with 100 mL of CH2Cl2, and the mixture was extracted two times with 50 mL of a saturated aqueous solution of NaHCO3. The organic phase was dried over Na2SO4. The solution was purified by column chromatography on silica gel with ethyl acetate/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) to give the product 5 as pale yellow (1.2 g, yield: 91%). 1H NMR (400 MHz, CDCl3) δ (ppm) 4.42–4.30 (q, 4H, CH2OCO), 4.19–4.14 (t, 2H, CH2CH2OCO), 3.28–3.23 (t, 2H, N3CH2), 1.91 (s, 12H, C(CH3)2Br, 1.64–1.57 (m, 4H, OCH2CH2 and N3CH2CH2), 1.28 (br m, 17H, (CH2)7 and CH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 172.41, 170.99, 66.37, 65.62, 55.32, 51.48, 46.65, 30.66, 29.42, 29.41, 29.19, 28.83, 28.71, 28.52, 26.70, 25.86, 17.94. ESI-MS (M + Na)+C24H41Br2N3O6 theoretical: 648.13, experimental: 648.04.
:9) to give the product 5 as pale yellow (1.2 g, yield: 91%). 1H NMR (400 MHz, CDCl3) δ (ppm) 4.42–4.30 (q, 4H, CH2OCO), 4.19–4.14 (t, 2H, CH2CH2OCO), 3.28–3.23 (t, 2H, N3CH2), 1.91 (s, 12H, C(CH3)2Br, 1.64–1.57 (m, 4H, OCH2CH2 and N3CH2CH2), 1.28 (br m, 17H, (CH2)7 and CH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 172.41, 170.99, 66.37, 65.62, 55.32, 51.48, 46.65, 30.66, 29.42, 29.41, 29.19, 28.83, 28.71, 28.52, 26.70, 25.86, 17.94. ESI-MS (M + Na)+C24H41Br2N3O6 theoretical: 648.13, experimental: 648.04.
Synthesis of azide mid-functionalized poly(styrene) (PS–N3–PS) (6).
Into a 50 mL Schlenk tube, St (10 mL, 87 mmol), PMDETA (0.073 mL, 0.349 mmol), and compound 5 (0.109 g, 0.174 mmol) in 5 mL of anisole were added and the reaction mixture was degassed by three freeze–pump–thaw cycles and left under argon. CuBr (0.05 g, 0.349 mmol) was added to the solution under argon. The tube was subsequently placed in a thermostatted oil bath at 80 °C for 60 min. After the specified time, the polymerization mixture was cooled in an ice bath and subsequently diluted with THF, passed through an alumina column to remove the catalyst, and two times precipitated in 100 mL cold methanol. The polymer was dried for 24 h under vacuum to afford PS–N3–PS as a white solid (0.54 g) ([M]0/[I]0 = 250, [I]0:[CuBr]0:[PMDETA]0 = 1:2:2). Mn,NMR = 3600 Da, Mn,SEC = 3800 Da, PDI = 1.08.
Synthesis of Hamilton wedge (HW) mid-functionalized poly(styrene) (PS–HW–PS) (7).
PS
–N
3
–
PS (0.4 g, 0.11 mmol), compound 12 (0.2 g, 0.33 mmol), copper (II) sulfate pentahydrate (0.08 g, 0.33 mmol) and sodium ascorbate (0.07 g, 0.33 mmol) were dissolved in DMF (10 mL). The resulting mixture was stirred at ambient temperature for 24 h before the coppercatalyst was removed by passing through a short column of neutral alumina. The solvent was removed under reduced pressure, subsequently diluted with the addition of DCM and extracted with EDTA solution to remove Cu, which is complexed by the recognition unit.22 The organic phase was dried over Na2SO4, concentrated and then two times precipitated in 100 mL methanol, filtered and dried under vacuum for 24 h to obtain a white solid (0.45 g, yield: 98%). Mn,NMR = 4200 Da, Mn,SEC = 4600 Da, PDI = 1.04. 1H NMR (400 MHz, CD2Cl2) δ 7.96–7.76 (9H, ArH of HW), 7.01–6.39 (5H, ArH of PS), 5.23–5.03 (2H, OCH2 linked to triazole), 3.45 (3H, CH3–O), 1.78–1.18 (aliphatic protons of PS).
Synthesis of CA end-functionalized poly(styrene) (PS–CA) (9).
Into a 50 mL Schlenk tube, St (8.0 mL, 53 mmol), PMDETA (0.073 mL, 0.26 mmol), and compound 8 (0.13 g, 0.26 mmol) in 2 mL of anisole were added and the reaction mixture was degassed by three freeze–pump–thaw cycles and left under argon. CuBr (0.05 g, 0.26 mmol) was added to the solution under argon. The tube was subsequently placed in a thermostatted oil bath at 110 °C for 45 min. The polymerization mixture was diluted with THF and passed through an alumina column to remove the catalyst. The solvent was removed under reduced pressure and subsequently diluted with the addition of DCM and extracted with EDTA solution to remove Cu, which is complexed by the recognition unit. The organic phase was dried over Na2SO4, concentrated and then two times precipitated in 100 mL methanol. The polymer was dried for 24 h in a Schlenk line to give a white solid (0.43 g) ([M]0/[I]0 = 200, [I]0:[CuBr]0:[PMDETA]0 = 1:1:1). Mn,NMR = 3600 Da, Mn,SEC = 3700 Da, PDI = 1.04. 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 2H of cyanuric acid), 7.01–6.39 (5H, ArH of PS), 4.42–4.33 (1H, CHBr), 3.73 (2H, CH2–N), 3.45 (2H, CH2–O), 1.78–1.18 (aliphatic protons of PS), 0.90–077 (6H, NCH2(CH2)3CH2O).
Synthesis of CA end-functionalized poly(methyl methacrylate) (PMMA–CA) (10).
Into a 50 mL Schlenk tube, MMA (5.0 mL, 46 mmol), PMDETA (0.048 mL, 0.23 mmol), and compound 8 (0.087 g, 0.23 mmol) in 5 mL of toluene were added and the reaction mixture was degassed by three freeze–pump–thaw cycles and left under argon. CuCl (0.023 g, 0.23 mmol) was added to the solution under argon. The tube was subsequently placed in a thermostatted oil bath at 50 °C for 30 min. The polymerization mixture was diluted with THF and passed through an alumina column to remove the catalyst. The solvent was removed under reduced pressure, subsequently diluted with the addition of DCM and extracted with EDTA solution to remove Cu, which is complexed by the recognition unit. The organic phase was dried over Na2SO4 and two times precipitated in 100 mL n-hexane. The polymer was dried for 24 h in a Schlenk line to give a white solid (0.78 g) ([M]0/[I]0 = 200, [I]0:[CuCl]0:[PMDETA]0 = 1:1:1). Mn,NMR = 9000 Da, Mn,SEC = 8500 Da, PDI = 1.13. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 2H of cyanuric acid), 4.02 (brs, 2H, –CH2OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 3.88 (brs, 2H, –NCH2–), 3.60 (brs, –OCH3 of PMMA), 1.78–1.18 (aliphatic protons of PMMA).
), 3.88 (brs, 2H, –NCH2–), 3.60 (brs, –OCH3 of PMMA), 1.78–1.18 (aliphatic protons of PMMA).
Synthesis of HW end-functionalized poly(styrene) (PS–HW) (13).
PS
–N
3 (1.00 g, 0.2 mmol), compound 12 (0.36 g, 0.61 mmol), copper (II) sulfate pentahydrate (0.15 mg, 0.61 mmol) and sodium ascorbate (0.12 mg, 0.61 mmol) were dissolved in DMF (5 mL). The resulting mixture was stirred at ambient temperature for 24 h before the coppercatalyst was removed by passing through a short column of neutral alumina. The solvent was removed under reduced pressure, subsequently diluted with the addition of DCM and extracted with EDTA solution to remove Cu which is complex by the recognition unit. The organic phase was dried over Na2SO4, concentrated and then two times precipitated in 100 mL methanol, filtered and dried under vacuum at 25 °C for 24 h to obtain a white solid (1.07 g, yield: 96%). Mn,NMR = 5500 Da, Mn,SEC = 5600 Da, PDI = 1.04. 1H NMR (400 MHz, CD2Cl2) δ 7.96–7.76 (9H, ArH of host), 7.01–6.39 (5H, ArH of PS), 5.23–5.03 (2H, OCH2 linked to triazole), 3.45 (3H, CH3–O), 1.78–1.18 (aliphatic protons of PS).
Self-assembly study between PS–HW and PS–CA.
A sample was prepared with dissolved PS–HW (11 mg, 2 × 10−3 mmol) and PS–CA (7.2 mg, 2 × 10−3 mmol) in 1 mL CD2Cl2 in an NMR tube. The mixture was kept at 40 °C for 5 min and then left to assemble overnight (12 h) at ambient temperature; subsequently 1H NMR spectra were recorded.
Self-assembly study between PS–HW–PS and PS–CA.
A sample was prepared with dissolved PS–HW–PS (8.4 mg, 2 × 10−3 mmol) and PS–CA (7.2 mg, 2 × 10−3 mmol) in 1 mL CD2Cl2 in an NMR tube. The mixtures were kept at 40 °C for 5 min and then left to assemble overnight (12 h) at ambient temperature; subsequently 1H NMR spectra were recorded.
Self-assembly study between PS–HW and PMMA–CA.
A sample was prepared with dissolved PS–HW (11 mg, 2 × 10−3 mmol) and PMMA–CA (18 mg, 2 × 10−3 mmol) in 1 mL CD2Cl2 in an NMR tube. The mixtures were kept at 40 °C for 5 min and then left to assemble overnight (12 h) at ambient temperature; subsequently 1H NMR spectra were recorded.
Self-assembly study between PS–HW–PS and PMMA–CA.
A sample was prepared with dissolved PS–HW–PS (8.4 mg, 2 × 10−3 mmol) and PMMA–CA (18 mg, 2 × 10−3 mmol) in 1 mL CD2Cl2 in an NMR tube. The mixtures were kept at 40 °C for 5 min and then left to assemble overnight (12 h) at ambient temperature; subsequently 1H NMR spectra were recorded.
Results and discussion
The Hamilton wedge (HW) and cyanuric acid (CA) motifs, which were chosen as binding units due to their high self-assembly constant (Ka = 106 M−1) in CH2Cl2 at ambient temperature,20e were introduced into well-defined PS and PMMA as mid-chain (refer to Scheme 2) and terminal (refer to Scheme 1) functionalities. The PS–HW–PS building block was designed following the synthetic pathway outlined in Scheme 3.
 |
| | Scheme 3 Synthetic strategy for preparing Hamilton wedge mid-chain functionalized poly(styrene) (PS–HW–PS (7)). | |
To incorporate a HW motif into a mid-chain position, PS–N3–PS was synthesized in several steps. The first involves reaction of 11-bromoundecan-1-ol with 2,2,5-trimethyl-1,3-dioxane-5-carboxylic acid to yield the corresponding compound 3. The 1H NMR spectrum of 3 shows the characteristic signals of –CH2OCO and –C–CH2O–C– appearing at 4.18 and 3.62 ppm, respectively (see Fig. S1 in the ESI†). In a second step, the deprotection of the acetonidegroup of compound 2 was readily accomplished in the presence of 1 M HCl. The two hydroxylgroups of 3 can readily be observed at 2.45 ppm and the signal for the methyl group of the acetonide functionality is no longer identified at 1.39–1.43 ppm (see Fig. S2 in the ESI†). In a third step, the bromide functionality of 3 was almost quantitatively converted to an azidegroup in the presence of NaN3. The desired azide compound was isolated in quantitative yields and used without further purification in the subsequent step. The 1H NMR spectrum of compound 4 (see Fig. S3 in the ESI†) indicates that the signals of –CH2Br and –CH2CH2Br at 3.41 ppm and 1.8 ppm disappeared and a new signal associated with –CH2N3 emerged at 3.28 ppm. The ATRPinitiator5 was obtained via esterification of 4 with 2-bromoisobutyryl bromide in the presence of triethylamine. The initiator was purified viacolumn chromatography and its purity was determined by 1H NMR and 13C NMR spectroscopy (see Fig. S5 in the ESI†). The 1H NMR spectrum of compound 5 (see Fig. S4 in the ESI†) indicates that the signals of –CH2–OH disappeared at 3.9–3.7 ppm and new signals associated with –CH2OCO and –CCH3Br were observed at 4.40 ppm and 1.91 ppm, respectively. In a final step, compound 5 was employed as an initiator for the ATRP of St in the presence of a CuBr/PMDETA complex system as catalyst in anisole to yield polymer6 (Mn = 3700 g mol−1, PDI = 1.06). Finally, utilizing copper catalyzed azide–alkyneconjugation chemistry, PS–N3–PS and compound 12 were reacted to give the corresponding PS–HW–PS in the presence of CuSO4 and sodium ascorbate in DMF at ambient temperature (see Fig. S10 in the ESI†). The number average molecular weight, Mn, of PS–HW–PS was determined viaSEC (molecular weight reported relatively to PS standards). The Mn of PS–HW–PS deduced viaSEC and 1H NMR reads 4600 Da (PDI = 1.04) and 4200 Da, respectively. The number average molecular weights as well as the corresponding polydispersity values of all of the above compounds are collated in Table 1.
Table 1 Characteristics of functional precursor polymers employed in the current study
|
Polymers
|
Monomer |
[M0]/[I0] |
Initiator
|
Time/min |
M
n
|
M
w/Mn |
M
n
|
|
[I]0/[PMDETA]0/[CuBr]0 = 1:2:2; at 80 °C.
[I]0/[PMDETA]0/[CuBr]0 = 1:1:1 at 110 °C.
[I]0/[PMDETA]0/[CuCl]0 = 1:1:1; at 50 °C.
[I]0/[PMDETA]0/[CuBr]0 = 1:1:1 at 110 °C.
Determined from RI detectionSEC using linear PS standards.
Determined from RI detectionSEC using linear PMMA standards.
Determined from 1H NMR spectroscopy.
|
|
6
|
St |
250 |
5
|
60 |
3700e |
1.06 |
3600 |
|
7
|
— |
— |
— |
— |
4600e |
1.04 |
4200 |
|
9
|
St |
200 |
8
|
45 |
3700e |
1.04 |
3600 |
|
10
|
MMA |
200 |
8
|
30 |
8500f |
1.13 |
9000 |
|
11
|
St |
200 |
MiBBr
|
45 |
4700e |
1.09 |
4900 |
|
13
|
— |
— |
— |
— |
5400e |
1.06 |
5500 |
After the successful synthesis of PS–HW–PS, CA end-functionalized PS (PS–CA), CA end-functionalized PMMA (PMMA–CA), and HW end-functionalized PS (PS–HW) were prepared (see Scheme 4) to serve as blocks for the homopolymer, diblock copolymer, star and miktoarm star polymers.
 |
| | Scheme 4 Preparation of CA- and HW-end-functionalized PS (PS–CA (9)) and PS–HW (13) and CA end-functionalized PMMA (PMMA–CA (10)). | |
PS
–CA was synthesized using compound 8 as an initiator for the ATRP of St in the presence of CuBr/PMDETA. Moreover, PMMA–CA was synthesized using 8 as an initiator during the ATRP of MMA in the presence of CuCl/PMDETA. Furthermore, PS–N3 was reacted with compound 12 to yield PS–HW through azide–alkyneconjugation employing the CuSO4/ascorbatecatalyst system in DMF at ambient temperature. The 1H NMR spectra (see Fig. S7, S8 and S9 in the ESI†) of PS–CA, PMMA–CA and PS–HW display all characteristic signals for the HW and CA moieties which were desired as end-group functionalities. The molecular weights of PS–CA (9), PMMA–CA (10) and PS–HW (13) were determined via1H NMR spectroscopy in addition to SEC. The SEC traces depicted in Fig. 1 confirm that the polymers were appropriately prepared with controlled molecular weight and low PDI. Table 1 summarizes the molecular weights of the polymers depicted in Schemes 3 and 4.
Before discussing in detail the self-assembly between the individual recognition motifs, CA and HW, it is appropriate to discuss the NMRsolvents, which are to be used to achieve self-assembly. Initially, the 1H NMR spectrum of PS–HW was recorded in CDCl3 and non-specific interactions between 13 ppm and 9 ppm (peaks labeled x, y, and z) were observed that can be attributed to the intermolecular H-bonding between the Hamilton receptors (see Fig. S6 in the ESI†) (see endnote [43] in ref. 29 for a discussion of such effects). The same measurement was repeated for PS–HW–PS and similar results were obtained. However, 1H NMR spectra of PS–HW and PS–HW–PS measured in CD2Cl2 evidence that the signals of the NH protons of the Hamilton receptor appear at 7.80 ppm and 8.52 ppm, which is in good agreement with theoretical expectations20t (see Fig. S7 and S10 in the ESI†). Moreover, it should be pointed out that the more polar solventCD2Cl2 solvates the Hamilton receptor better than CDCl3 and prevents the formation of inter/intra-molecular H-bonding. Thus, the polarity of CD2Cl2 is sufficient to prevent inter- and intramolecular H-bonding of the Hamilton receptors, yet appropriate to enable intermolecular H-bonding between HW and CA. On the basis of the above experimental data, CD2Cl2 is to be preferred over CDCl3 as NMRsolvent for investigating the self-assembly behavior between these recognition motifs.
Subsequently, the formation of diblock, star, and miktoarm star polymersvia H-bonding between the HW receptor and the CA located at various places of the polymer backbone (as described in Schemes 1 and 2) was investigated. All self-assembly processes of the building blocks can be followed by monitoring characteristic changes in the chemical shifts of the NHprotons of the two recognition pairs using 1H NMR spectroscopy. First, the formation of a PS–HW·PS–CA (PS–PS) self-assembled structure between PS–HW and PS–CA in CD2Cl2 at ambient temperature was studied. Upon the addition of 1 equiv. of PS–CA into 1 equiv. of PS–HW in CD2Cl2, it is observed that the 1H NMR spectrum (see Fig. 2) reveals strong shifts of the amideprotons (Hb and Hc) of the Hamilton receptor from 7.80 and 8.52 ppm (see Fig. S7 in the ESI†) to 9.64 and 9.16 ppm, respectively, thus evidencing that the self-assembled homopolymerPS–HW·PS–CA (PS–PS) is formed.
 |
| | Fig. 2 Expanded 1H NMR spectrum of the PS–HW·PS–CAself-assembly system in CD2Cl2 at ambient temperature. Equimolar amounts with respect to the HW and CA binding motifs were employed. The individual end group concentration for both motifs was close to 2 mM. | |
Furthermore, a new signal appears at 13.03 ppm (Ha) that corresponds to the (dynamically) bound cyanuric acid unit in the self-assembly motif.20t The yield of supramolecular homopolymer can subsequently be deduced from the ratio between the peak area of –CHBr as an ω-end of PS–CA at 4.55–4.42 ppm and the peak area of bound NH as an α-end of PS–CA at 13.03 ppm: the amount of total chains carrying a cyanuric acid end group is directly proportional to the signal intensity of –CHBr, whereas the amount of these chains bound to the Hamilton receptor is directly proportional to the new NH signal at 13.03 ppm. Therefore—considering that the NHproton occurs with double intensity—the relative number of cyanuric acid terminal chains bound in the (dynamic) self-assembly at any point in time can be derived. The percentage of self-assembled polymer was thus deduced close to ∼89% (see Fig. 3 for the individual signals with their respective integral values). Moreover, during the addition of 1 equiv. of PMMA–CA to 1 equiv. of PS–HW in CD2Cl2, it is observed that the 1H NMR spectrum (see Fig. 4) also reveals strong respective shifts of the amideprotons (Hb and Hc) of the Hamilton receptor from 7.80 and 8.52 ppm (see Fig. S7, ESI†) to 9.36 and 9.80 ppm, respectively, thus evidencing that a supramolecular block copolymerPS–HW·PMMA–CA (PS–PMMA) is formed.
 |
| | Fig. 3
1H NMR spectrum of the PS–HW·PS–CA supramolecular polymer system in CD2Cl2 at ambient temperature with the integral values of the relevant resonances. From the ratio of the peak areas a to d, the degree of self-assembly can be deduced as being close to ∼89%. Equimolar amounts with respect to the HW and CA binding motifs were employed. The individual end group concentration for both motifs was close to 2 mM. | |
 |
| | Fig. 4 Expanded 1H NMR spectrum of the PS–HW·PMMA–CAself-assembly system in CD2Cl2 at ambient temperature. Equimolar amounts with respect to the HW and CA binding motifs were employed. The individual end group concentration for both motifs was close to 2 mM. | |
Furthermore, a new (broad) signal20t appears at 12.16 ppm (Ha) that corresponds to the bound cyanuric acid unit in the self-assembled structure. In contrast to the PS–HW·PS–CA system, the percentage of self-assembled polymer cannot be calculated from the 1H NMR spectrum in this case due to the absence of a separate, non-overlapping peak to compare to the bound protons. The absence of a suitable comparative resonance is due to the methyl group present in methyl methacrylate, in contrast to the poly(styrene) system. In addition, no other resonances associated with the chain terminus are available for integration without overlap. However, it is reasonable to assume that the supramolecular block copolymer, PS–HW·PMMA–CA (PS–PMMA), was formed in high yield similar to supramolecular miktoarm star polymer, (PS)2PMMA (see below), because 1H NMR spectra of the two self-assembly systems display almost the same chemical shifts of the imideprotons in the Hamilton wedge (in PS–PMMA: 9.36 and 9.80 ppm and in (PS)2PMMA: 9.32 and 9.79 ppm).
As noted above, the formation of supramolecular star polymersviaself-assembly between PS–HW–PS and PS–CA was also examined. When 1 equiv. of PS–HW–PS was mixed with 1 equiv. of PS–CA in CD2Cl2 at ambient temperature, the chemical shift of the imideprotons of PS–CA changed from 7.85 (see Fig. S9 in the ESI†) to 13.05 ppm and the NHproton of the PS–HW–PS appeared at 9.64 and 9.14 ppm due to the self-assembly (see Fig. 5). The yield of supramolecular three arm star polymer was calculated from the ratio between the peak area of –CH2CH2–triazole of PS–HW–PS at 4.24 ppm and the peak area of bound NH as an α-end of PS–CA at 13.05 ppm, employing an identical rationale for arriving at the degree of self-assembly as described and employed above. The percentage of self-assembled star polymerPS3 was thus calculated to be close to 90% by 1H NMR spectroscopy (see Fig. 6 for the integrals of the individual peak assignments). In principle, it should also be possible to use the –CHBr proton (as in the PS–HW·PS–CA case discussed above) to assess the degree of binding within the miktoarms star system. However, using the methyleneprotons next to the triazole system is in principle the preferred option (if feasible), as due to the ATRP process not all chains may carry a bromide terminus. In the case of the PS–HW·PS–CA system, this option is not available, as no non-overlapping protons in the vicinity of the triazole system are present. It should be noted, however, that the chain end fidelity (i.e. the percentage of chains featuring both a cyanuric acid and bromide terminus) is close to 98% (calculated from the integral areas of the cyanuric acid imide and –CHBr protons) in the PS–CA precursor macromolecule.24
 |
| | Fig. 5 Expanded 1H NMR spectrum of the PS–HW–PS·PS–CAself-assembly system in CD2Cl2 at ambient temperature. Equimolar amounts with respect to the HW and CA binding motifs were employed. The individual end group concentration for both motifs was close to 2 mM. | |
 |
| | Fig. 6
1H NMR spectrum of the PS–HW–PS·PS–CA supramolecular star polymer system in CD2Cl2 at ambient temperature. From the ratio of the peak areas a to d (integral values shown), the degree of self-assembly can be deduced as being close to ∼90%. Equimolar amounts with respect to the HW and CA binding motifs were employed. The individual end group concentration for both motifs was close to 2 mM. | |
Finally, the preparation of a miktoarm star polymerPS–HW–PS·PMMA–CA (PS2PMMA) was investigated through the self-assembly between PS–HW–PS and PMMA–CA. After the addition of 1 equiv. of PMMA–CA to 1 equiv. of PS–HW–PS in CD2Cl2, it is observed that the 1H NMR spectrum (see Fig. 7) reveals similar strong shifts of the amideprotons (Hb and Hc) of the Hamilton receptor from 7.80 and 8.52 ppm (see Fig. S10 in the ESI†) to 9.32 and 9.79 ppm, respectively, thus once more evidencing that a self-assembly—in this case a miktoarm star polymerPS–HW–PS·PMMA–CA (PS2PMMA)—is formed. Furthermore, a new broad signal appears at 12.42 ppm (Ha) that corresponds to the bound cyanuric acid unit in the self-assembly. Following the above established calculation routine, the yield of supramolecular miktoarm star polymer was calculated from the ratio between the peak area of –CH2CH2–triazole of PS–HW–PS at 4.24 ppm and the peak area of bound NH as an α-end of PMMA–CA at 12.42 ppm. The percentage of self-assembled star polymerPS2PMMA was calculated to be close to 98% by 1H NMR spectroscopy (see Fig. 8 for the integrals of the individual peak assignments). Similar to the above discussed homo-three armed star system, the deduction of the degree of binding via the –CHBr proton is the less preferred option (although it provides results within approximately similar range).
 |
| | Fig. 7 Expanded 1H NMR spectrum of the PS–HW–PS·PMMA–CAself-assembly system in CD2Cl2 at ambient temperature. Equimolar amounts with respect to the HW and CA binding motifs were employed. The individual end group concentration for both motifs was close to 2 mM. | |
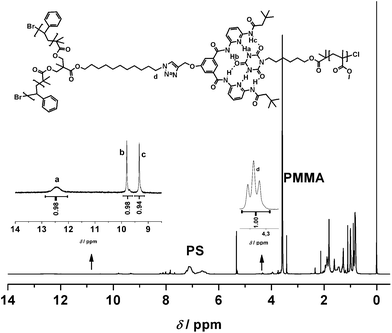 |
| | Fig. 8
1H NMR spectrum of the PS–HW–PS·PMMA–CA supramolecular miktoarm star polymer system in CD2Cl2 at ambient temperature with integrated values from the ratio of the peak areas a to d. The degree of self-assembly can be deduced as being close to ∼98%. Equimolar amounts with respect to the HW and CA binding motifs were employed. The individual end group concentration for both motifs was close to 2 mM. | |
Conclusions
Well-defined narrow polydispersity polymers containing a terminal and mid-chain functional Hamilton wedge as well as cyanuric acid terminal motifs function efficiently during the self-assembly into block and star (co)polymer structures. The Hamilton wedge and cyanuric acid functional macromolecular building blocks were prepared via a combination of atom transfer radical polymerization and copper catalyzed azide–alkyne ligation. Provided the binding motif decorated prepolymer systems carry a suitable unit allowing for the identification of individual chain terminal protons, the self-assembly process can be studied at ambient temperature viaproton NMR spectroscopy, evidencing very high degrees of binding in two cases in excess of 90%, attesting the almost quantitative formation of block copolymer structures in CH2Cl2. In addition, we have evidenced that the six hydrogen bonds between the Hamilton wedge and cyanuric acid offer stronger binding compared to other systems such as the three-hydrogen bond forming diaminopyridine–thymineassembly. This in turn offers a better option for the preparation of self-assembled polymers with variable topologies. The present data thus indicate that self-assembly driven macromolecular design is readily achievable employing Hamilton wedge and cyanuric acid motifs based on ATRPpolymers at ambient temperatures.
Acknowledgements
C.B.-K. is grateful for continued support from the Karlsruhe Institute of Technology (KIT) in the context of the Excellence Initiative as well as by the German Research Council (DFG) and the Ministry of Science and Arts of the State of Baden-Württemberg. O.A. thanks the Islamic Development Bank (IDB) for a PhD scholarship. The authors gratefully acknowledge stimulating discussions with Prof. B. Luy (KIT) on NMR spectroscopy.
References and notes
-
J. Roovers, in Star and Hyperbranched Polymers, ed. M. Mishra and S. Kobayashi, Marcel Dekker, New York, 1998, p. 285 Search PubMed.
- W. Burchard, Adv. Polym. Sci., 1999, 143, 113–194 CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS.
- A. Hirao, M. Hayashi, S. Loykulnant, K. Sugiyama, S. W. Ryu, N. Haraguchi, A. Matsuo and T. Higashihara, Prog. Polym. Sci., 2005, 30, 111–182 CrossRef CAS.
-
(a) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS;
(b) C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS;
(c) M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS;
(d) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS;
(e) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS;
(f) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS;
(g) J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS;
(h) F. Di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef CAS;
(i) V. Percec and B. Barboiu, Macromolecules, 1995, 28, 7970–7972 CrossRef CAS;
(j) B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171–1185 RSC.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
-
(a) M. J. Krische and J.-M. Lehn, Struct. Bonding (Berlin, Ger.), 2000, 96, 3–29 Search PubMed;
(b) F. H. Beijer, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 1998, 37, 75–78 CrossRef CAS.
-
(a) R. Knapp, A. Schott and M. Rehahn, Macromolecules, 1996, 29, 478–480 CrossRef CAS;
(b) J. P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez and C. Coudret, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS;
(c) M. Chiper, M. A. R. Meier, D. Wouters, S. Hoeppener, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Macromolecules, 2008, 41, 2771–2777 CrossRef CAS.
-
(a) C. Nuckolls, T. J. Katz, G. Katz, P. J. Collings and L. Castellanos, J. Am. Chem. Soc., 1999, 121, 79–88 CrossRef CAS;
(b) L. Brunsveld, E. W. Meijer, R. B. Prince and J. S. Moore, J. Am. Chem. Soc., 2001, 123, 7978–7984 CrossRef CAS.
-
(a) N. Yamaguchi and H. W. Gibson, Angew. Chem., Int. Ed., 1999, 38, 143–147 CrossRef CAS;
(b) P. R. Ashton, P. J. Campbell, E. J. T. Chrystal, P. T. Glink, S. Menzer, D. Philp, N. Spencer, J. F. Stoddart, P. A. Tasker and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1995, 34, 1865–1869 CrossRef CAS.
- J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
-
(a) O. Ikkala and G. Brinke, Science, 2002, 295, 2407 CrossRef CAS;
(b) A. V. Ambade, C. Burd, M. N. Higley, K. P. Nair and M. Weck, Chem.–Eur. J., 2009, 15, 11904–11911 CrossRef CAS;
(c) S. K. Yang, A. V. Ambade and M. Weck, Chem.–Eur. J., 2009, 15, 6605–6611 CrossRef CAS;
(d) C. R. South, C. Burd and M. Weck, Acc. Chem. Res., 2007, 40, 63–74 CrossRef CAS;
(e) W. B. Binder, M. J. Kunz, C. Kluger, G. Hayn and R. Saf, Macromolecules, 2004, 37, 1749–1759 CrossRef CAS;
(f) C. Burd and M. Weck, Macromolecules, 2005, 38, 7225–7230 CrossRef CAS;
(g) W. H. Binder, S. Bernstorff, C. Kluger, L. Petraru and M. J. Kunz, Adv. Mater., 2005, 17, 2824–2828 CrossRef CAS;
(h) D. Lu, Y. Wang, T. Wu, K. Tao, L. An and R. Bai, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5805–5815 CrossRef CAS;
(i) F. Huang, D. S. Nagvekar, S. Slebodnick and H. W. Gibson, J. Am. Chem. Soc., 2005, 127, 484–485 CrossRef CAS;
(j) A. V. Ambade, S. K. Yang and M. Weck, Angew. Chem., Int. Ed., 2009, 48, 2894–2898 CrossRef CAS;
(k) E. B. Berda, J. E. Foster and E. W. Meijer, Macromolecules, 2010, 43, 1430–1437 CrossRef CAS;
(l) S. H. M. Sontjens, R. A. E. Renken, G. M. L. van Gemert, T. A. P. Engels, A. V. Bosman, H. M. Janssen, L. E. Govaert and F. P. T. Baaijens, Macromolecules, 2008, 41, 5703–5708 CrossRef CAS;
(m) S. Chen, A. Bertrand, X. Chang, P. Alcouffe, C. Ladaviere, J.-F. Gerard, F. Lortie and J. Bernard, Macromolecules, 2010, 43, 5981–5988 CrossRef CAS;
(n) X. Yang, F. Hua, K. Yamato, E. Ruckenstein, B. Gong, W. Kim and C. Y. Ryu, Angew. Chem., Int. Ed., 2004, 43, 6471–6474 CrossRef CAS;
(o) K. E. Feldman, M. J. Kade, T. F. A. de Greef, E. W. Meijer, E. J. Kramer and C. J. Hawker, Macromolecules, 2008, 41, 4694–4700 CrossRef CAS;
(p) O. Altintas, I. Yilmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3242–3249 CrossRef CAS;
(q) A. D. Celiz and O. A. Scherman, Macromolecules, 2008, 41, 4115–4119 CrossRef CAS;
(r) B. G. G. Lohmeijer and U. S. Schubert, Angew. Chem., Int. Ed., 2002, 41, 3825–3829 CrossRef CAS;
(s) D. J. M. van Beek, M. A. J. Gillissen, B. A. C. van As, A. R. A. Palmans and R. P. Sijbesma, Macromolecules, 2007, 40, 6340–6348 CrossRef CAS;
(t) S. K. Yang, A. V. Ambade and M. Weck, J. Am. Chem. Soc., 2010, 132, 1637–1645 CrossRef CAS.
- E. M. Todd and S. C. Zimmerman, J. Am. Chem. Soc., 2007, 129, 14534–14535 CrossRef CAS.
-
(a) A. Likhitsup, S. Yu, Y.-H. Ng, C. L. L. Chai and E. K. W. Tam, Chem. Commun., 2009, 4070–4072 RSC;
(b) Y. Shen, H. Tang and S. Ding, Prog. Polym. Sci., 2004, 29, 1053–1078 CrossRef CAS.
- J. Bernard, F. Lortie and B. Fenet, Macromol. Rapid Commun., 2009, 30, 83–88 CrossRef CAS.
- O. Altintas, P. Gerstel, N. Dingenouts and C. Barner-Kowollik, Chem. Commun., 2010, 46, 6291–6293 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- S. K. Chang and A. D. Hamilton, J. Am. Chem. Soc., 1988, 110, 1318–1319 CrossRef CAS.
- H. Ihre, A. Hult, J. M. J. Frechet and I. Gitsov, Macromolecules, 1998, 31, 4061–4068 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6458–6465 CrossRef CAS.
- V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J. M. Lehn, Chem.–Eur. J., 2002, 8, 1227–1244 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.