A fluorescent all-fluorene polyazomethine—towards soluble conjugated polymers exhibiting high fluorescence and electrochromic properties†
Satyananda
Barik
and
W. G.
Skene
*
Laboratoire de caractérisation photophysique des matériaux conjugués, Department of Chemistry, Pavillon JA Bombardier, University of Montreal, CP 6128, succ, Centre-ville, Montreal, Quebec, Canada H3T 2B1. E-mail: w.skene@umontreal.ca
First published on 7th February 2011
Abstract
A conjugated all-fluorene polyazomethine (6) was prepared from the condensation of 9,9′-dioctyl fluorene dialdehyde and 9,9′-dihexyl fluorene diamine. The resulting polymer was soluble in common organic solvents courtesy of the solubilizing alkyl groups in the 9,9′ positions. Determination of the polymer's molecular weight was subsequently possible using standard characterization methods. Polymer formation was confirmed both by GPC and NMR with average number molecular weights ranging from 9 to 145 kg mol−1. The absolute fluorescence quantum yield (Φfl) of 6 in dichloromethane was 0.19 while in thin films the value increased to 0.40. The fluorescence could further be restored to unity at 77 K while Φfl = 0.76 was possible by protonating 6 with trifluoroacetic acid. 6 underwent stark color changes from yellow to red upon doping with TFA and FeCl3 and the resulting intermediates could be neutralized with Et3N and hydrazine hydrate, respectively. Reversible protonation/deprotonation was possible both in solution and thin films. Meanwhile, electrochemical oxidation of 6 at ca. 1.5 V resulted in a 120 nm bathochromic change in the absorbance. The intermediate could be reduced at −0.2 V to regenerate the neutral 6 that absorbed at 425 nm. The Job method confirmed that the oxidized intermediate was the radical cation.
Introduction
Conjugated polyfluorenes and their derivatives have attracted a great deal of interest for use in organic light emitting diodes (OLEDs). This is in part owing to their inherent high fluorescence yield that is well suited for such emitting applications. Although fluorescent polymers have been successfully prepared using Yamamato, Stille, Suzuki, Gilch and Horner–Emmons coupling protocols,1–5 these require stringent reaction conditions. The resulting polymers must further be extensively purified to remove undesired by-products for obtaining pristinepolymers required for preparing efficient emitting materials with high color purity. Alternate straightforward coupling methods for preparing conjugated polyfluorenes not requiring stringent protocols with simple- to no-product purification are therefore advantageous, providing that the opto-electronic properties are not compromised.An alternate method for conjugated polymer preparation is the use of azomethine (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) connections. These are highly attractive alternatives to vinylene linkages in part owing to their ease of preparation, not requiring stringent reaction conditions.6 They are further advantageous because the main by-product is water, therefore they can be isolated with little- or no purification. Moreover, the azomethine bond is isoelectronic to its all-carbon counterpart. Polyazomethines are therefore ideally suited for functional materials usage given their comparable opto-electronic properties to their homologues with the advantage of simple preparation and little purification.7–9
C–) connections. These are highly attractive alternatives to vinylene linkages in part owing to their ease of preparation, not requiring stringent reaction conditions.6 They are further advantageous because the main by-product is water, therefore they can be isolated with little- or no purification. Moreover, the azomethine bond is isoelectronic to its all-carbon counterpart. Polyazomethines are therefore ideally suited for functional materials usage given their comparable opto-electronic properties to their homologues with the advantage of simple preparation and little purification.7–9
Despite these advantages, azomethine derivatives of fluorenepolymers have not been pursued as functional materials for emitting devices. This is a result of poor performance and incompatible properties of previously investigated fluorenyl polyazomethine derivatives for use in devices. In particular, previously investigated fluoreneazomethines were nonfluorescent, a property which is of critical importance for OLED usage.10,11 This is further compounded with limited solubility in common solvents making optimal structure/performance and accurate property assessment difficult.
The inadequate properties of polyazomethines for device usage have resulted in little attention being given to azomethine property improvement. This is particularly true for the fluorescence especially since polyazomethines are generally understood to be nonfluorescent regardless of structure. Polyazomethines are further understood to be hydrolytically unstable and decompose under acidic conditions. As a result, little effort has been devoted for preparing conjugated polyazomethines that fluoresce in amounts comparable to their carbon counterparts. We were therefore incited to prepare a fluorescent polyfluorene azomethine for demonstrating that the inherent fluorescence of fluorophore containing polyazomethines could be restored by suppressing their otherwise fluorescence quenching processes. This was further encouraged by our previous studies demonstrating that the fluorescence of azomethine model compounds could be significantly increased to detectable amounts by simple structural modification and temperature.12–14 We were further motivated to prepare a conjugated polyfluorene azomethine that was soluble in common organic solvents for polymer characterization using standard techniques. This is of importance given the limited solubility of previously investigated polyazomethines making their characterization challenging.10,11,15 Molecular weight characterization using standard methods is further essential for accurate polyazomethine molecular weight–property evaluation and for confirming polymer formation. To satisfy this means, a soluble conjugated polyfluorene azomethine was prepared. The opto-electronic properties including electrochemical and chemical oxidation of the highly fluorescent and soluble polyazomethine are herein described. These properties are for illustrating the robustness of the azomethine bonds and that functional material properties including fluorescence and electrochromics are possible with such polyazomethines.
Materials and methods
Reagents and solvents were received from commercial sources and were used as received unless otherwise stated. Anhydrous and deoxygenated solvents were obtained with an activated alumina column system. 1H and 13C NMR spectra were recorded at room temperature on a 400 MHz spectrometer. All samples were dissolved in deuterated solvents and the spectra were referenced to the solvent line (CDCl3: 1H, 7.26 and 13C 77.0 ppm) relative to TMS.Spectroscopic measurements
Absorption measurements were done on a Cary-500 spectrometer and fluorescence studies were carried out on an Edinburgh Instruments FLS-920 fluorimeter after deaerating the samples thoroughly with nitrogen for 20 minutes. Relative fluorescence quantum yields were measured at 10−5 M by exciting the corresponding compounds at its maximum absorption in spectroscopic grade methylcyclohexane relative to itself at both 77 K and room temperature under the same conditions. Absolute quantum yields were done using an integrating sphere that was calibrated to fluorescein in ethanol.16Doping of 6 was done in dichloromethane (10−5 M) and stock solutions of FeCl3 and hydrazine were sequentially added (5 μL) to the polymer solution and the resulting absorbance spectrum recorded. Similarly, stock solutions of trifluoroacetic acid (TFA) and triethylamine (TEA) were sequentially added (5 μL) to the polymer solution and the resulting absorbance spectrum recorded. For thin film doping studies, a 10 mg mL−1 solution of 6 was filtered with a 0.45 μm PTFE filter and then spin coated onto washed glass slides at 2000 rpm for 30 seconds followed by increasing the speed to 5000 rpm. The spin coated samples were protonated using TFA vapour and then neutralized by dipping the glass slide into a solution of TEA diluted in dichloromethane. Similarly, the Job method for determining the oxidation stoichiometry was done using stock solutions of known mole fractions (1.0 mM) of both 6 and ferric chloride.
Electrochemical measurements
Cyclic voltammetry measurements were performed on a Bio Analytical Systems EC Epsilon potentiostat. Compounds were dissolved in anhydrous and deaerated dichloromethane at 10−4 M with 0.3 M TBAF. A platinumelectrode and a saturated Ag/AgCl electrode were employed as auxiliary and reference electrodes, respectively.Molecular weight measurements
GPC analyses were performed on a Waters Breeze system equipped with a 717 plus autosampler, a 1525 Binary HPLCpump and a 2410 refractive index detector. Three Styragel columns HR3, HR4 and HR6 (7.8 × 300 mm) in series were used for resolving the different samples. The flow rate of the THFeluent was 1 mL min−1. The temperature of the columns was 33 °C. Molecular weights were calculated using a polystyrene kit SM-105 (10 standards) from Shodex.Thermal and calorimetric analyses
Thermogravimetric analysis (TGA) studies were carried out on a Hi-Res TGA 2950 thermogravimetric analyser (TA Instruments) with a 10 °C min−1 ramp to 700 °C (Tdec was defined as the onset of the decomposition temperature). Differential scanning calorimetry (DSC) studies were carried out on a TA instrument with a 5 °C min−1 ramp from −50 to 220 °C under both N2 and ambient atmospheres.Syntheses
The syntheses of 1–4 and 7–10 were prepared according to reported procedures.17–19Polyfluorenylazomethine (6)
Step-growthpolymerization was performed by combining 2 (200 mg, 0.55 mmol) and 3 (245 mg, 0.55 mmol) in CHCl3 (10 mL) in a pressure tube. A catalytic amount of diluted TFA (60 μL) was then added (Scheme 1). The pressure tube was sealed and heated to 90 °C for 124 h. The reaction mixture was cooled to room temperature and it was then poured into methanol to afford the title product as a yellow solid. The solid was filtered and washed with methanol and water followed by acetone and then dried under vacuum overnight to afford the desired polymer (380 mg, 80%). 1H NMR (400 MHz, CDCl3,) δ ppm = 10.1 (m, CHO), 8.68 (m, NCH–), 7.70–7.78 (m, Fl–H), 2.03 (m, –CH2), 1.07 (m, –CH2–CH2–), 0.68 (m, –CH3). Various molecular weights as seen in Table 1 were possible with various heating times.
 | ||
| Scheme 1 Polymerization scheme for 6. | ||
| Sample | Polymerization time/h | M n/g mol−1 | M w/g mol−1 | PDI |
|---|---|---|---|---|
| a Relative to polystyrene standards. | ||||
| i | 48 | 9400 | 12 600 | 1.35 |
| ii | 96 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
21300 |
1.28 |
| iii | 124 | 145800 |
192500 |
1.32 |
Results and discussions
Synthesis and characterization
Instead of using commercially available diaminofluorene as the monomer for preparing the targeted polyfluorene azomethine (6), the dialkylated 2 was chosen. This was because the resulting polymer was expected to be soluble in commonly used characterization solvents such as THF. Further motivation for preparing 6 from 2 was that the resulting polymer was expected to be fluorescent based on the knowledge gained from our previous azomethine photophysical studies of model compounds.12,13,17,18,20,21 We were further incited to prepare 6 from 2 because this fully alkylated polymer had never been prepared and hence investigated. The mono-substituted monomer analogues were desired for benchmarking the opto-electronic properties and for comparing to 6. All the products were confirmed by 1H and 13C NMR and mass spectrometry.Given that 6 was expected to be soluble in standard organic solvents, its preparation was done using our previously successful polymerization method.22 Exact stoichiometric amounts of 2 and 3 were combined and then refluxed in chloroform with a catalytic amount of TFA. This straightforward polymerization method is in contrast to previously used polymerization conditions for azomethine preparation that involved dehydration reagents such as LiCl, high boiling point solvents, and TiCl4, all of which require stringent reaction protocols and extensive purification for isolating the desired polymer. In contrast, pristine 6 was obtained by simple removal of the solventin vacuo after polymerization. Alternatively, the polymer could be precipitated from methanol. Conversely, purification by soxhlet extraction for 24 hours was required for the previously investigated 5.11 Therefore, the polymerization of 6 is advantageous relative to other polyazomethine fluorene derivatives owing in part to the straightforward condensation protocol that does not require stringent reaction conditions and extensive postpolymerization purification.10,23
The obtained 6 was soluble in THF, chloroform, dichloromethane, in addition to other standard organic solvents. Characterization using standard methods including GPC and NMR was therefore possible. The GPC (Fig. 1) spectrum shows the molecular weight evolution with polymerization time and the corresponding GPC data in Table 1 confirm that 6 is indeed a polymer. This is in contrast to previously reported derivatives such as 5 that consisted of low molecular weight oligomers that were only sparingly soluble in hot NMP and DMAC.24 Further evidence for the polymeric nature of 6 is had from its 1H NMR spectrum (see ESI†). The salient features of the NMR spectrum are the lack of discrete resolvable peaks, consistent with polymers and not oligomers, while the imineprotons seen at 8.7 ppm confirm formation of the azomethine bond. The resolvable imine and aldehyde peaks allow the Mn to be approximately calculated from the ratio of two discrete peaks and the calculated value is consistent with the GPC data. Meanwhile, the multiple aldehyde peaks are consistent with a polydisperse polymer measured by GPC.
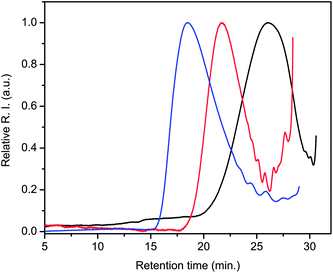 | ||
| Fig. 1 GPC chromatograms of 6 illustrating the evolution of molecular weight as a function of polymerization time 48 (black), 96 (red) and 124 (blue) hours of polymerization at 90 °C. | ||
The thermal decomposition of 6 was determined by thermogravimetric analysis (TGA) under nitrogen at 10 °C min−1. As seen in Fig. 2, the polymer decomposes at 380 °C. The decomposition temperature was consistent when heating under both nitrogen and ambient atmospheres. The high decomposition temperature confirms the thermal robustness of the polymer, and particularly, the heteroatomic bond. Meanwhile, a glass transition temperature of 130 °C was determined by differential scanning calorimetry (inset, Fig. 2) under nitrogen at 5 °C min−1 for the 145 kg mol−1 sample of 6.
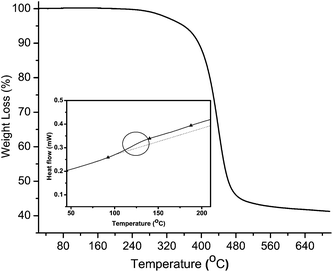 | ||
| Fig. 2 Thermal analysis of 6 (Mn = 16.6 kg mol−1) measured at 10 °C min−1 under N2. Inset: DSC curve of 6 measured at 5 °C min−1 under N2. | ||
Fluorescence properties
The absorbance and fluorescence spectra of 6 in both solution and thin films are shown in Fig. 3. It is noteworthy that both the spectroscopic and electrochemical properties were independent of molecular weight. From the figure, it is evident that both the absorbance and fluorescence in thin films are bathochromically shifted relative to those measured in solution. This is in part due to the increased coplanarization of the fluorene–azomethine moieties that are otherwise twisted by up to 65°.18,19 The bathochromic shift is further a result of interchain interactions. Meanwhile, the absorbance of 6 is bathochromically shifted by 40 nm relative to polyfluorene implying a greater degree of conjugation concomitant with electronic effects resulting from the electron withdrawing azomethine.5 The high degree of conjugation of 6 is further confirmed by its absorbance and fluorescence spectra that are bathochromically shifted relative to its low molecular weight counterpart 5.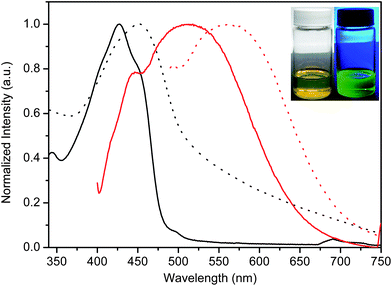 | ||
Fig. 3 Normalized absorbance (black) and fluorescence (red) spectra of 6 (Mn = 145.8 kg mol−1) in dichloromethane (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and thin film (⋯). Inset: images of 6 in dichloromethane under ambient light (left) and irradiated with at 325 nm UV lamp (right). ) and thin film (⋯). Inset: images of 6 in dichloromethane under ambient light (left) and irradiated with at 325 nm UV lamp (right). | ||
The fluorescence yield (Φfl) of 6 is of particular interest, especially given that previously investigated conjugated polyazomethines do not fluoresce regardless of structure.10,15,22,25–27 It should be noted that quantum yields are usually referenced against known compounds. The challenge with this approach is that both the reference and compound of study must absorb and fluoresce at approximately the same wavelength in addition to fluorescing the same amount. Large errors in Φfl often result because either one or both of the actinometric criteria are not met. We therefore measured the Φfl using an integrating sphere. This allows for accurate and absolute emission yield measurements independent of the excitation and emission wavelengths and quantum yields, providing that Φfl ≥ 0.05. Additionally, this method does not require a reference. As seen in Table 2, 6 fluoresces significantly relative to its corresponding monomers. In fact, the fluorescence can be visually detected to the naked eye as shown in the inset of Fig. 3. The measured value of Φfl =0.19 for 6 is in contrast to its unalkylated analogue 5 whose Φfl < 0.01. It is obvious that alkylation plays an important role in enhancing the fluorescence. However, alkylation is expected to decrease the fluorescence since fluorene fluorescence is known to be deactivated by electronic effects and substitution.13,28 This is evident for monomers 1–4 that do not fluoresce compared to pristine monomeric fluorene, whose Φfl = 0.72 (Chart 1).18,19
| Compound | Thin-film | Φ fl (H+)a | E pa /eV | E pc /eV | HOMOd/eV | LUMOd/eV | E g elec/eVe | |
|---|---|---|---|---|---|---|---|---|
| λ max/nm | λems/nm | |||||||
| a Absolute fluorescence quantum yield at room temperature in dichloromethane. The value in parentheses is the fluorescence quantum yield upon protonation at room temperature. b Oxidation potential relative to saturated Ag/Ag+electrode. c Reduction potential relative to Ag/Ag+. d Relative to the vacuum level. e Electrochemical energy gap. f Reliable electrochemical data for 1 and 2 could not be obtained as a result of their instability under our experimental conditions. g From ref. 10 and 11. h From ref. 11, 29, 30, 33, and 34. | ||||||||
| 1 | 292 | 389 | 0 | — | — | — | — | — |
| 2 | 398 | 494 | 0.01 | — | — | — | — | — |
| 3 | 339 | 383 | 0 | 1.0 | −1.2 | 5.4 | 3.2 | 2.2 |
| 4 | 326 | 366 | 0.01 | 1.3 | −1.2 | 5.7 | 3.2 | 2.5 |
| 5 | 436 | 456 | <0.01 | 1.1 | −1.4 | 5.2 | 2.7 | 2.5 |
| 6 | 452 | 563 | 0.19 (0.75) | 1.5 | −1.7 | 5.9 | 2.7 | 3.2 |
| Polyfluoreneh | 379 | 415 | 0.42–0.60 | 1.59 | −2.16 | −5.50 | 2.37 | 3.13 |
 | ||
| Chart 1 Monomers and polyfluorene azomethine prepared and examined and a representative analogue. | ||
The substantial fluorescence of 6 relative to 5, implies that aminofluorenealkylation suppresses nonradiative deactivation modes that are otherwise responsible for fluorescence quenching. The observed fluorescence of 6 further suggests that polyazomethine fluorescence quenching occurs predominately by rotation around the azomethine–fluorene bonds. The aminofluorenealkylation increases the rotation barrier such that energy dissipation by this deactivation pathway is no longer efficient at room temperature. The fluorescence yield in thin films of 6 was subsequently measured to confirm this energy dissipation mode since deactivation pathways involving bond rotation are further hindered in this state. The absolute fluorescence measured (Φfl = 0.4) in spin coated films confirmed that rotation around the fluorenyl–azomethine bond rotation is predominately responsible for deactivating the singlet excited state of polyazomethines. Although the value measured in thin film is comparable to polyfluorene prepared by Suzuki or Yamamoto coupling methods, other fluorescence deactivation modes are nonetheless present, accounting for the remaining 60% loss in fluorescence.29–31
Low temperature fluorescence of 6 was done to see whether the fluorescence could be further increased. At 77 K, all deactivation pathways involving bond rotation and vibration are completely suppressed. The fluorescence of 6 increased to near unity at 77 K, confirming that deactivation by rotation/vibration deactivation modes other than rotation around the fluorene–azomethine bond are also present. The 60% fluorescence quenching observed in thin films is most likely a result of energy dissipation occurring possibly via vibration/rotation of the 9,9′-fluorenealkyl groups. These are known to be efficient fluorescence deactivation pathways for alkylated oligo- and polyfluorene.12,32
A substantial increase in fluorescence yield was also possible in solution by protonating the azomethine bond with TFA. Although the exact mechanism for fluorescence increase with protonation is unknown, the fluorescence of 6 can nonetheless be turned-on to achieve emission yields greater than its polyfluorene counterpart (Table 2). The collective fluorescence data confirm that the polyazomethines can be made fluorescent with judicious choice of substitution in addition to modulating the temperature and by protonating the azomethine. Compound 6 is thus the first example of a fluorescent conjugated polyazomethine that is not only fluoresces significantly, but whose emission can also be modified by temperature and protonation.
Azomethines are understood to be hydrolytically unstable, and as a result, they decompose in the presence of acid. The robustness of the azomethine is spectroscopically demonstrated via repeated protonation/deprotonating. As seen in Fig. 4, the protonation of 6 with TFA results in a 90 nm bathochromic shift in absorbance. The resulting intermediate can subsequently be deprotonated with TEA to regenerate the neutral 6. The corresponding color change induced by protonation and deprotonation is shown in the lower inset of Fig. 4. It is noteworthy that the absorbance spectrum of the neutral 6 after having been exposed to multiple TFA/TEA cycles is identical to that of pristine 6. The consistently regenerated absorbance spectrum of 6 after protonation/deprotonation cycles confirms the robustness of the azomethine. The same intermediate was also obtained when oxidizing 6 with FeCl3. Similar to the neutralization with TEA, the intermediate could be reduced with hydrazine hydrate to regenerate the neutral 6 (upper inset, Fig. 4). The polymer could also withstand repeated chemical doping/dedoping cycles with FeCl3/hydrazine without significant spectral changes, further confirming the robustness of the polymer.
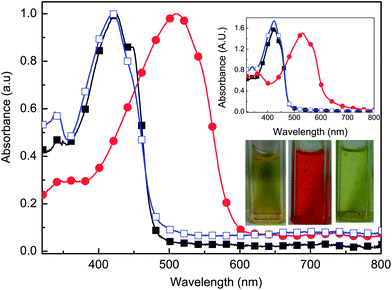 | ||
| Fig. 4 Absorbance spectra of 6 (■; Mn = 9.4 kg mol−1), with TFA added (●), followed by the addition TEA (□) in dichloromethane. Upper inset: absorbance spectra of 6 (■), oxidation with FeCl3 (●), followed by neutralization with hydrazine hydrate (□) in dichloromethane. Lower inset: colour change of 6 (left) upon addition of TFA (middle) followed by the addition of TEA (right) in dichloromethane. | ||
To further examine the chemical robustness of the polyazomethine, its doping/dedoping in thin films was examined. The polymer was spin coated onto glass slides and the samples were exposed to TFA vapour. The film color changed from yellow to orange upon immediate exposure to the acid vapour. After a given period of time, the protonated glass slide was submerged into a dichloromethane solution of TEA. The corresponding absorbance spectra are shown in Fig. 5. As in the case of solution doping, multiple cycles of doping/dedoping of the thin film could be done without significant changes in the absorbance spectra. The polyazomethine therefore can be reversibly doped/dedoped without decomposition in both solution and thin films and serve to illustrate the robustness of the azomethines towards hydrolysis and oxidative decomposition.
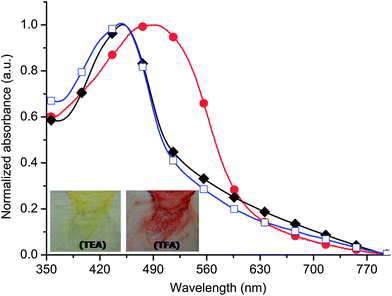 | ||
| Fig. 5 Absorbance of 6 (Mn = 9.4 kg mol−1) in thin-films (■), exposed to TFA vapour (●), followed by submerging the sample in a dichloromethane solution of TEA (□). Inset: images of color change upon exposing the glass substrate coasted with a thin film of 6 to TFA vapour followed by TEA. | ||
Electrochemical properties
Cyclic voltammetry (CV) was done to asses the electrochemical properties and the energy levels of 6 and its corresponding monomers. Unfortunately, electrochemical data for 1 and 2 could not be obtained owing to their instability under our experimental conditions. The monomers 3 and 4 in addition to 6 exhibited both oxidation and reduction processes (Fig. 6), confirming their p- and n-type behavior. The onsets of these potentials were used to calculate the HOMO and LUMO energy levels in addition to the energy-gap (Eg) according to known methods.35 The calculated values are reported in Table 2. The large redox potentials measured further confirm the robustness of the azomethine. However, a less positive oxidation potential was expected for 6 given its important degree of conjugation. The observed value suggests that the polymer is not fully conjugated, must likely owing to known twisting along the backbone. This is supported by crystallographic studies demonstrating that the fluorenyl moiety is twisted by up to 65° from the azomethine bond to which it is connected.12,19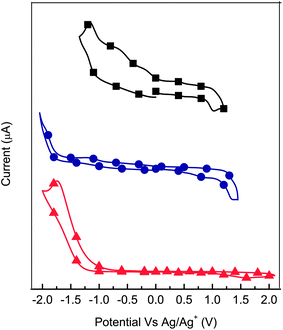 | ||
| Fig. 6 Cyclic voltammetry of 3 (■), 4 (●), and 6 (▲; Mn = 9.4 kg mol−1) in dichloromethane at a scan rate of 100 mV s−1. | ||
Spectroelectrochemical properties
The spectroelectrochemistry of 6 was investigated for assessing the spectroscopic properties of the electrochemically produced intermediate. This was done with an electrochemical cell consisting of a mesh Ptworking electrode, reference electrode (Ag/Ag+) and counter electrode (Pt-wire). Both the working and counter electrodes were placed in a UV-viscell with a narrow path length and the cell was placed in the sample light beam of the spectrophotometer. Upon applying increasing potentials from 0 to 1.5 V, a stark color change from yellow to red occurred with 6 (Fig. 7). Upon electrochemical oxidation, the initial yellow color of the neutral 6 (λmax = 425 nm) bleached concomitant with the formation of a new peak at 520 nm corresponding to a red color. Complete conversion of 6 to its corresponding radical cation occurred after applying a potential above 1.4 V. Meanwhile, the yellow color of the neutral 6 could be restored upon reducing the red radical cation at ca. −0.2 V. The obvious isosbestic point seen at 462 nm taken together with reversible absorbances at 425 and 520 nm confirms the lack of polymerdecomposition and further serve to illustrate the robustness of the polyfluorenylazomethine towards oxidative decomposition.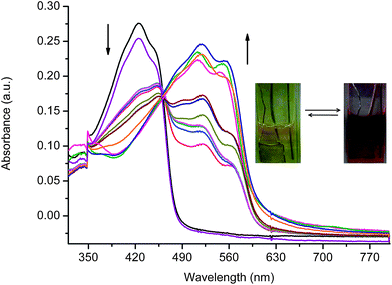 | ||
| Fig. 7 Spectral change of 6 (Mn = 145.8 kg mol−1) in dichloromethane at a scan rate of 100 mV s−1 with increasing applied potentials ranging between 0 and 1.5 V vs.Ag/Ag+ and neutralization with −0.2 V (purple line). Inset: photographs of neutral (left) and oxidized (right) 6 after applying 0/1.5 and −0.2 V, respectively. | ||
The Job method was subsequently used for determining the stoichiometry of the FeCl3/6oxidation process and for assigning the nature of the electrochemically induced intermediate. This was possible by measuring the change in absorbance as a function of FeCl3 mole fraction. It is evident from Fig. 8 that the chemical oxidation stoichiometry is 1:1. The fact that FeCl3 is a one-electron oxidant taken together with the measured 1:1 stoichiometry, it can be concluded that the chemically produced intermediate is a radical cation. The similar absorbance of both the anodically produced intermediates by chemical and electrochemical means confirms that the radical cation is generated in both cases.
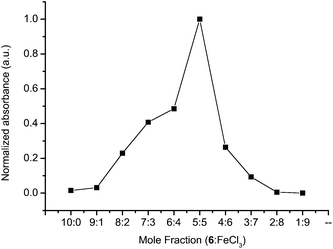 | ||
| Fig. 8 Job plot of 6 (Mn = 145.8 kg mol−1) with FeCl3 in dichloromethane monitored at 520 nm. | ||
Conclusion
A highly conjugated fluorene polyazomethine soluble in standard organic solvents was successfully prepared and characterized. Polymer formation was confirmed both by GPC and NMR. Of particular interest is the polymer's fluorescence that could be switched-on as a result of alkylation of the diaminofluorene moiety. The fluorescence yield was doubled in thin film and was restored to quantitative amounts at 77 K, demonstrating that azomethine fluorescence quenching predominately occurs by bond rotation/vibration processes. Similar fluorescence enhancement to near unity was possible upon protonating with TFA. Not only is this the first example of a highly fluorescent conjugated polyazomethine, but it also demonstrates that polyazomethine fluorescence is contingent on structure. Moreover, the intrinsic fluorescence behaviour of fluorophores incorporated into polyazomethines can be preserved by suppressing otherwise fluorescence quenching bond rotation processes, possible with structural modification. Meanwhile, reversible chemical doping with TFA/Et3N and FeCl3/hydrazine couples epitomizes the robustness of conjugated azomethines towards decomposition. The stark color change between the neutral and doped states of 6 observed both in solution and thin film confirms the sensor like properties possible with conjugated polyazomethines and demonstrates that they posses electrochromic properties. Functional materials and sensor properties are therefore possible with polyazomethines. Their ease of synthesis and little purification compared to their carbon analogues make polyazomethines interesting potential replacements for materials currently used for opto-electronic applications.Acknowledgements
The authors acknowledge financial support from the Natural Sciences and Engineering Research Council Canada for both a DG and SRG in addition to the Centre for Self-Assembled Chemical Structures and additional equipment funding from the Canada Foundation for Innovation. Mr S. Eissenbaum's assistance with the TGA and DSC measurements is appreciated. WGS also thanks both the Alexander von Humboldt Foundation and the RSC for a JWT Jones Travelling fellowship, allowing the completion of this manuscript.References
- E. Scheler and P. Strohriegl, J. Mater. Chem., 2009, 19, 3207–3212 RSC.
- Y. Chen, F. Li and Z. Bo, Macromolecules, 2010, 43, 1349–1355 CrossRef CAS.
- N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110–1119 CrossRef CAS.
- N. Yu, R. Zhu, B. Peng, W. Huang and W. Wei, J. Appl. Polym. Sci., 2008, 108, 2438–2445 CrossRef CAS.
- B. Lui, W.-L. Yu, J. Pei, S.-Y. Liu, Y.-H. Lai and W. Huang, Macromolecules, 2001, 34, 7932–7940 CrossRef CAS.
- S. A. Pérez Guarìn, M. Bourgeaux, S. Dufresne and W. G. Skene, J. Org. Chem., 2007, 72, 2631–2643 CrossRef.
- C. Wang, S. Shieh, E. LeGoff and M. G. Kanatzidis, Macromolecules, 1996, 29, 3147–3156 CrossRef CAS.
- C.-J. Yang and S. A. Jenekhe, Chem. Mater., 1991, 3, 878–887 CrossRef CAS.
- J. E. G. Kuder, W. Harry and D. Wychick, J. Org. Chem., 1975, 40, 875–879 CrossRef CAS.
- F.-C. Tsai, C.-C. Chang, C.-L. Liu, W.-C. Chen and S. A. Jenekhe, Macromolecules, 2005, 38, 1958–1966 CrossRef CAS.
- C.-L. Liu and W.-C. Chen, Macromol. Chem. Phys., 2005, 206, 2212–2222 CrossRef CAS.
- S. Dufresne, S. A. P. Guarìn, A. Bolduc, A. N. Bourque and W. G. Skene, Photochem. Photobiol. Sci., 2009, 8, 796–804 RSC.
- S. Dufresne, I. U. Roche, T. Skalski and W. G. Skene, J. Phys. Chem. C, 2010, 114, 13106–13112 CrossRef CAS.
- S. Dufresne, T. Skalski and W. G. Skene, Can. J. Chem., 2011 DOI:10.1139/cjc10153 , in press.
- S.-H. Jung, T.-W. Lee, Y. C. Kim, D. H. Suh and H. N. Cho, Opt. Mater., 2003, 21, 169–173 CrossRef CAS.
- W. R. Ware and W. Rothman, Chem. Phys. Lett., 1976, 39, 449–453 CrossRef CAS.
- S. Barik, S. Friedland and W. G. Skene, Can. J. Chem., 2010 Search PubMed , in press.
- D. Tsang, M. Bourgeaux and W. G. Skene, J. Photochem. Photobiol., A, 2007, 192, 122–129 CrossRef CAS.
- S. A. Pérez Guarìn, S. Dufresne, D. Tsang, A. Sylla and W. G. Skene, J. Mater. Chem., 2007, 17, 2801–2811 RSC.
- S. Dufresne, L. Callaghan and W. G. Skene, J. Phys. Chem. B, 2009, 13, 15541–15549 CrossRef.
- A. N. Bourque, S. Dufresne and W. G. Skene, J. Phys. Chem. C, 2009, 113, 19677–19685 CrossRef CAS.
- M. Bourgeaux and W. G. Skene, Macromolecules, 2007, 40, 1792–1795 CrossRef CAS.
- D. Sek, B. Jarzabek, E. Grabiec, B. Kaczmarczyk, H. Janeczek, A. Sikora, A. Hreniak, M. Palewicz, M. Lapkowski, K. Karon and A. Iwan, Synth. Met., 2010, 160, 2065–2076 CrossRef CAS.
- The DPn = 4 calculated from the data available from ref. 11.
- C.-L. Liu, F.-C. Tsai, C.-C. Chang, K.-H. Hsieh, J.-L. Lin and W.-C. Chen, Polymer, 2005, 46, 4950–4957 CAS.
- C.-P. Chang, C.-C. Wang, C.-Y. Chao and M.-S. Lin, J. Polym. Res., 2005, 12, 1–7 Search PubMed.
- J.-S. K. Hyun-Chul Kim, K.-S. Kim, H.-K. Park, S. Baek and M. Ree, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 825–834 CrossRef.
- R. S. Murphy, C. P. Moorlag, W. H. Green and C. Bohne, J. Photochem. Photobiol., A, 1997, 110, 123–129 CrossRef CAS.
- C. Xia and R. C. Advincula, Macromolecules, 2001, 34, 5854–5859 CrossRef CAS.
- H. P. M. OliveiraI, T. D. MartinsI, K. M. HonórioII, P. C. RodriguesII, L. AkcelrudII, A. B. F. SilvaIII and T. D. Z. Atvars, J. Braz. Chem. Soc., 2009, 20, 160–166.
- J. M. Kauffman, P. T. Litak, J. A. Novinski, C. J. Kelley, A. Ghiorghis and Y. Qin, J. Fluoresc., 1995, 5, 295–305.
- K. L. Chan, M. Sims, S. I. Pascu, M. Ariu, A. B. Holmes and D. D. C. Bradley, Adv. Funct. Mater., 2009, 19, 2147–2154 CrossRef CAS.
- M. Ariu, D. G. Lidzey, M. Sims, A. J. Cadby, P. A. Lane and D. D. C. Bradley, J. Phys.: Condens. Matter, 2002, 14, 9975 CrossRef CAS.
- F.-K. Su, J.-L. Hong and L.-L. Lin, J. Appl. Polym. Sci., 2007, 106, 3308–3314 CrossRef CAS.
- J. r. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0py00394h |
| This journal is © The Royal Society of Chemistry 2011 |