Grafting mixed responsive brushes of poly(N-isopropylacrylamide) and poly(methacrylic acid) from gold by selective initiation
Xiaofeng
Sui
a,
Szczepan
Zapotoczny
ab,
Edmondo M.
Benetti
ac,
Mine
Memesa
a,
Mark A.
Hempenius
a and
G. Julius
Vancso
*a
aMaterials Science and Technology of Polymers, MESA+ Institute for Nanotechnology, University of Twente, Enschede, NL-7500, The Netherlands. E-mail: g.j.vancso@utwente.nl; Fax: +31 53 489 3823; Tel: +31 53 489 2967
bFaculty of Chemistry, Jagiellonian University, Ingardena 3, PL-30-060, Krakow, Poland
cLaboratory for Surface Science and Technology, Department of Materials, ETH Zurich, Wolfgang-Pauli-Strasse 10, CH-8093, Zürich, Switzerland
First published on 28th January 2011
Abstract
Mixed polymer brushes consisting of poly(N-isopropylacrylamide) (PNIPAM) and poly(methacrylic acid) (PMAA) were prepared at room temperature by a sequential combination of surface-initiated atom transfer radical polymerization (SI-ATRP) and iniferter-mediated photopolymerization (SI-IMP). Mixed monolayers of disulfides of ATRP initiator and IMP iniferter on gold substrates were employed for independent initiation of the two polymer components. As the polymerization of PNIPAM and PMAA was performed in successive steps, the chain length and the corresponding brush height of the constituents could be individually controlled. Grazing angle Fourier transform infrared spectroscopy (FTIR) and ellipsometry were performed to monitor the component-specific formation of the grafts. The responsive behavior of the mixed polymer brushes under different pH was investigated by contact angle (CA) measurements, atomic force microscopy (AFM) and in situellipsometry. Reversible structural reorganization and variation of brush thickness and of wetting characteristics were monitored.
1. Introduction
Responsive films made of surface-tethered macromolecules have been widely applied to a variety of substrates and used to control surface wetting and friction, in fabrication of micro- and nanostructures, for preparing actuators, for regulation of cell and protein attachment and for many other purposes.1–11 These responsive films with well-defined structural properties are usually obtained by controlled surface-initiated radical polymerization methods.12 The surface properties of stimuli responsive homopolymer brushes usually exhibit sharp transitions between e.g. two different physical states in response to a small change of an external “environmental” trigger (e.g. temperature, pH or light). In mixed brushes, it is possible to switch the surface performance between the properties of the two different polymer components, which allows for control in a broader range of the targeted property if compared to the single constituents.13–19Two different strategies are generally used to synthesize mixed polymer brushes: the so-called “grafting-to” and “grafting-from” techniques.8 In the first approach, binary brush platforms can be obtained by sequential attachment of two different polymers onto reactive substrates.20,21As an example, this specific strategy allowed the surface grafting of polybases and polyacids within the same brush film. The obtained mixed polyelectrolyte brushes were characterized by the surface charge polarity and density which could be precisely tuned by immersion in media presenting different pH values.22 Despite its broad applications, “grafting-to” approaches present several limitations, i.e. relatively low grafting densities and film thicknesses are usually achieved by this preparation method. Hence, in situgrowth of polymer brushes from pre-functionalized surfaces by a “grafting-from” approach is usually preferred. Surface-initiated polymerization from immobilized initiator adsorbates was used in several instances to obtain mixed brushes with adjustable thicknesses and high grafting densities. The mixed monolayer approach is attractive since it is possible to immobilize two or more initiators that may be activated independently. After the first polymer is grafted, the film may be cleaned and reactivated under different conditions in order to synthesize the second grafted polymer. The net result is a mixed brush where two kinds of polymer chains are intercalated between each other.23–26
The preparation and the stimuli-responsive behavior of PNIPAM and PMAA homopolymer, random copolymer and block copolymer brushes were described in some reports.27,28 However, to our knowledge, the preparation and the responsive behavior of PNIPAM–PMAA mixed brush films have not been investigated. PNIPAM and PMAA are often employed in responsive systems, hence we chose these two constituents for this study.
In this work, we combined two surface-initiated controlled radical polymerization techniques, i.e. surface-initiated atom transfer radical polymerization (SI-ATRP) and iniferter-mediated photopolymerization (SI-IMP), to create mixed homopolymer brushes of PNIPAM and PMAA on gold substrates at room temperature. We chose SI-ATRP to synthesize the first brush component (PNIPAM) and subsequently SI-IMP for the second brush (PMAA). The particular step-wise fabrication of ATRP and IMP was selected as they allowed us to sequentially control brush synthesis by the two different mechanisms of polymerization. This is ensured as activation of an ATRP initiator with a metal complex is a bimolecular process, while the free radicals in IMP are formed by photodecomposition, which is a unimolecular process. A unimolecular activation mechanism was preferred for the grafting of the second brush component due to the steric hindrance induced by the existing polymer chains. We also mention that SI-IMP was well suited for the direct synthesis of PMAA brushes since it alleviated the problems of catalyst complexation, dissociation, or disproportionation which could occur when ATRP of electrolytic monomers in protic media is performed.15,29
The choice of gold substrate presented several advantages over the use of frequently reported silicon surfaces. SAMs of disulfides/thiols on Au surfaces are well ordered, they can be easily formed under environmental conditions and their compositions may be easily varied by tuning the relative concentration of the different adsorbates in the feeding solutions.3,30–32
The brush synthesis used here is outlined in Scheme 1. The behavior of the mixed brush films was characterized and investigated in different media. In particular, the pH responsive behavior was monitored by contact angle measurements, AFM and in situellipsometry.
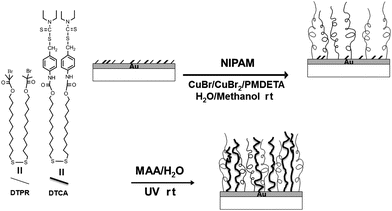 | ||
| Scheme 1 Synthetic strategy for mixed polymer brushes from immobilized precursors on gold by combining SI-ATRP and SI-IMP. | ||
2. Experimental section
2.1. Materials
N-Isopropylacrylamide (NIPAM, Aldrich, 97%) was recrystallized twice from a toluene/hexane solution (50%, v/v) and dried under vacuum prior to use. Methacrylic acid (MAA, Aldrich, 99%) was purified by vacuum distillation. Copper(I) bromide (CuBr, Aldrich, 98%) was purified by stirring in glacial acetic acid, filtering, and washing with ethanol three times, followed by drying in vacuum at room temperature overnight. Copper(II) bromide (Aldrich, ≥99%), methanol (Biosolve, absolute) and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (98%, Acros Organics) were used as received. The ATRP initiator, dithiodiundecane-11,1-diylbis[2-bromo-2-methylpropanoate] (DTPR, depicted in Scheme 1), was synthesized according to literature procedures.33 The photoiniferter, dithiodiundecane-11,1-diylbis[4-({[(diethylamino)carbonothioyl] thioethyl} phenyl)carbamate] (DTCA, depicted in Scheme 1), was synthesized following the procedure reported previously.3Gold substrates (200 nm thick Au layer) were obtained from Ssens BV, Hengelo, The Netherlands. Buffer solutions, pH 3.0 and pH 8.0, were obtained from Merck. Water was purified with a Millipore desktop system. All other chemicals were received from Aldrich and were used without further purification.2.2. Formation of mixed SAMs on gold substrates
Mixed SAMs were prepared by immersing gold substrates in chloroform solutions containing predetermined amounts of the ATRP initiator (DTPR) and of the photoiniferter (DTCA). The substrates were previously cleaned with “piranha” solution, and rinsed with water, ethanol and dichloromethane extensively. Caution: ‘Piranha’ solutions react violently with many organic materials and should be handled with great care. The total concentration of the adsorbates was 1 mM. Substrates covered with only one monolayer (DTPR or DTCA) were also prepared for reference experiments. The typical deposition time was 16 h. The substrates were rinsed sequentially with dichloromethane and ethanol, dried in a stream of N2 and immediately used for brush growth. For most of the reported polymerizations, the mixed SAMs were prepared from a solution containing DTPR and DTCA with a molar ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 in the feed.
:30 in the feed.
2.3. Synthesis of PNIPAM brushes by SI-ATRP
Scheme 1 displays the synthetic pathway for the preparation of PNIPAM brushes on Au. NIPAM (5.73 g, 50 mmol) monomer and PMDETA (314 μL, 1 mmol) were added to a water (18 mL) and methanol (2 mL) mixture. The solution was purged with argon for 30 min. CuBr (71.7 mg, 0.5 mmol) and CuBr2 (11.2 mg, 0.05 mmol) were added into another reaction flask and also flushed with argon. Monomer, ligand and catalyst were then combined and stirred for another 30 min to facilitate the formation of the organometallic complex. This solution was then transferred into the flasks containing the substrates covered with SAMs. The flasks were sealed with rubber septa and kept at room temperature under argon. After reaching the desired reaction time (30 min), the substrates were removed from the polymerization solution, exhaustively rinsed with water to remove any unreacted and not surface tethered substances and subsequently dried in a stream of nitrogen.2.4. Synthesis of PMAA polymer brushes by SI-IMP
The substrates were placed in flasks containing 1.0 M aqueous solution of MAA, they were extensively purged with argon and finally irradiated for the pre-determined polymerization time by an array of six UV-B lamps (15 W, G15T8E, Ushio, Japan; sample-to-lamp distance, 20 cm). Following completion of photopolymerization, the samples were extensively rinsed with water to remove all traces of the polymerization solution, and subsequently dried in a stream of nitrogen.2.5. Characterization
Static contact angle measurements with water were performed by the sessile drop technique using an optical contact angle device equipped with an electronic syringe unit (OCA15, Dataphysics, Germany). At least three different measurements of each sample were performed.X-Ray photoelectron spectroscopy (XPS) was used to evaluate the actual surface concentration in SAMs. XPS spectra were obtained by a Quantera XPS instrument (Physical Electronics) using monochromatized Al Kα radiation (1486.6 eV) with an X-ray beam diameter of 100 μm and a take-off angle of 30°, relative to the substrate's surface. Each sample was measured over a set of eight different locations. To evaluate the relative concentration of DTCA and DTPR on the surface, high-resolution XPS elemental scans for the O1s, N1s and Br3p signals were recorded. By combining the values obtained for each sample, the relative composition of the SAMs was determined.
Grazing angle FTIR spectroscopy was employed to determine the actual surface composition of the polymer brushes. FTIR spectra were obtained using a BIO-RAD FTS575C spectrometer equipped with a nitrogen-cooled cryogenic mercury telluride detector. A background spectrum was obtained by scanning a clean gold substrate. For reference purposes, a set of nine thin polymer films with known, different compositions of PNIPAM and PMAA homopolymer mixtures was prepared by drop casting from methanol solutions onto silicon substrates. FTIR spectra of these mixed homopolymer samples were used for calibration. The relative intensities of the deconvoluted peaks at 1730 cm−1 (characteristic for PMAA) and at 1650 cm−1 (characteristic for PNIPAM) were plotted against the known compositions of the films and a calibration curve was determined. The composition of the mixed brushes was derived from their FTIR spectra using the so-obtained calibration.
The thickness of dry brushes was measured using a rotating analyzer ellipsometer (J. A. Woollam VASE) at 10 nm intervals at wavelengths from 380 to 800 nm at an angle of incidence of 70°. The measurements were performed at 22 °C at 30% relative humidity. The thickness and refractive index determination were performed on at least 5 spots for each sample. In situ ellipsometric measurements were conducted at 22 °C with the same instrument using a quartz flow cell with thin walls fixed at an angle of 70° with respect to the sample plane. The angle of incidence of the light was set such that its path was normal to the window. The samples were immersed in different buffer solutions for at least 15 min to ensure the full swelling. The measured values of amplitude ratio upon reflection and phase shift were used to obtain the optical constants of the samples by the standard WVASE 32 software package (J. A. Woollam Co.).
AFM in the tapping mode (TM-AFM) was employed to study the morphology of the brush surfaces. A Dimension D3100 (AFM, Digital Instruments, Veeco-Bruker, Santa Barbara, CA) was applied.
3. Results and discussion
The preparation of mixed PNIPAM–PMAA brushes was carried out as summarized in Scheme 1. During the first step mixed SAMs were deposited on Au from chloroform solutions of mixtures of DTPR and DTCA. The formation of the SAMs was confirmed by FTIR (data not shown) and by XPS measurements. Quantitative determination of the surface composition was performed on the mixed SAMs as the feed composition and the surface composition can differ due to differences in the absorption kinetics of the individual components onto Au. The surface concentration of DTPR molecules as obtained by XPS was 37.5 mol%, which was lower than the value for the corresponding feed solution (70 mol%). This difference was attributed to the higher polarity of DTCA functionalities with respect to the DTPR, which makes the chemisorption of DTCA on the Au surface energetically favored.34–37Following deposition of the mixed initiating SAMs, PNIPAM was grafted from the functionalized Au surfaces by SI-ATRP. The formation of PNIPAM grafted layers was confirmed by ellipsometry and contact angle measurements. Ellipsometry demonstrated the formation of PNIPAM brush layers with a dry film thickness value of 64 ± 2 nm following 30 min polymerization time. The contact angle value of this brush was 56 ± 2°, which is typical for PNIPAM films.38 We note that recent studies showed that alkyl dithiocarbamate iniferters for IMP can also initiate ATRP in the presence of copper catalysts with nitrogen-based ligands. These studies showed that the initiation efficiency was strongly dependent on the molecular structure of the iniferter and on the reaction conditions. Also there was essentially no “halogen exchange” between the iniferter reagent and Br in the catalyst solution.39–44 In order to confirm the activity of only DTPR molecules as initiators for SI-ATRP from the mixed SAMs and the consequent inertness of DTCA adsorbates during ATRP, control experiments under the same polymerization conditions were performed using pure DTCA monolayers. We observed no formation of any grafted polymer. Pure DTPR SAMs stimulated the growth of brush layers with an average ellipsometric thickness of 155 ± 3 nm over a 30 min polymerization time.
To synthesize binary mixed PNIPAM/PMAA brushes with various PMAA chain lengths, samples from the same PNIPAM brush batch featuring 64 ± 2 nm dry height were exposed to photopolymerization conditions at room temperature for various polymerization times. The resulting thickness values were determined by ellipsometry and AFM (Fig. 1). As shown in Fig. 1, the dry brush thickness increased monotonically with the irradiation time. The thickness results obtained by ellipsometry were in good agreement with the height increments recorded by AFM in air. To confirm that PMAA chains were indeed initiated from the adsorbed photoiniferters, we exposed a pure ATRP initiator monolayer to the typical IMP conditions. After cleaning, no considerable changes were observed in the brush thickness confirming the high specificity of the DTCA molecules.
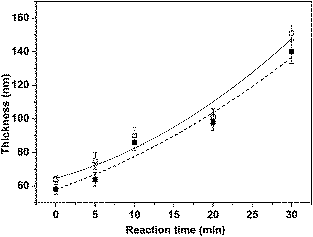 | ||
| Fig. 1 Film thickness vs.IMP polymerization time for the mixed brushes, starting with a PNIPAM brush, measured by ellipsometry (open squares, solid line) and AFM (filled squares, dashed line). The fitted lines are to guide the eye. | ||
It should be noted that the thickness of the PNIPAM layer synthesized from mixed monolayers is significantly smaller than the thickness of a PNIPAM layer synthesized from a pure DTPR SAM under the same time interval. This difference is most likely due to the lower initiator concentration.34,45The thickness of PMAA initiated from a PNIPAM layer is also smaller than the one obtained from a full DTCA monolayer. In this case, the lower growth rate is most likely caused by the limited diffusion of the monomer to the surface already partially covered by PNIPAM brushes.
FTIR analysis confirmed the presence of PMAA and PNIPAM immobilized on the surfaces of the substrates. In representative spectra shown in Fig. 2a, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching yields a strong band at 1650 cm−1, while the two bands at 1370 and 1390 cm−1 are assigned to the two methyl groups in the isopropyl functionality of PNIPAM. In Fig. 2e (one component PMAA brushes), the intense peak around 1730 cm−1is assigned to the CO stretching vibration in the carboxylic acid groups of PMAA. In addition, C–O stretching bands, characteristic to PMAA, can be observed at 1203 and 1269 cm−1. The FTIR spectra of the mixed brushes captured in Fig. 2b–d have characteristic peaks for both PMAA and PNIPAM. The increasing content of PMAA in the mixed brushes with the polymerization time was also verified. The ratio of the heights of the peak at 1730 cm−1 (PMAA) and the peak at 1650 cm−1 (PNIPAM) increased with the irradiation time.
O stretching yields a strong band at 1650 cm−1, while the two bands at 1370 and 1390 cm−1 are assigned to the two methyl groups in the isopropyl functionality of PNIPAM. In Fig. 2e (one component PMAA brushes), the intense peak around 1730 cm−1is assigned to the CO stretching vibration in the carboxylic acid groups of PMAA. In addition, C–O stretching bands, characteristic to PMAA, can be observed at 1203 and 1269 cm−1. The FTIR spectra of the mixed brushes captured in Fig. 2b–d have characteristic peaks for both PMAA and PNIPAM. The increasing content of PMAA in the mixed brushes with the polymerization time was also verified. The ratio of the heights of the peak at 1730 cm−1 (PMAA) and the peak at 1650 cm−1 (PNIPAM) increased with the irradiation time.
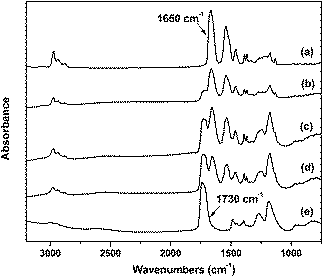 | ||
| Fig. 2 FTIR spectra of (a) PNIPAM homopolymer brush; (b–d) mixed polymer brushes: starting from the same height of PNIPAM, after various photopolymerization times for PMAA: (b) 5 min, (c) 10 min, (d) 20 min, and (e) PMAA homopolymer brush. | ||
The sample following 20 min of IMP of MAA (53 mol% of PMAA) was chosen to investigate the responsive behavior of binary PNIPAM/PMAA brush films (sample named as PNIPAM/PMAA-20 min).
PMAA undergoes a marked pH-induced conformational transition in aqueous solutions. At low pH values PMAA chains are weakly charged and adopt a collapsed conformation to prevent exposure of the hydrophobic methyl groups to the polymer–water interface. At high pH values the charge density along the PMAA chains markedly increases causing intramolecular repulsion and a subsequent stretching of the polymer chains.46
In situ
ellipsometry at room temperature (22 °C) shows that exposure of a dry PNIPAM/PMAA-20 min brush to the pH = 3 buffer solution swells the layer 1.5 times to its dry-layer thickness, while immersion in pH = 8 buffer solution results in a 3.0 times change as compared to the dry-layer thickness. For subsequent measurements, the PNIPAM/PMAA-20 min samples were immersed in a buffer solution with pH = 3 or 8 for 10 min. After each treatment, the samples were quickly rinsed with anhydrous ethanol, dried in a stream of nitrogen and immediately used for FTIR, AFM and contact angle analyses. FTIR spectra for this mixed brush and the reference homopolymer brushes that received the same treatments are shown in Fig. 3. FTIR spectra are very useful as the degree of protonation of the –COOH groups can directly be monitored (by assessing the –CO band at 1730 cm−1 for –COOH stretching, in comparison with the band at 1570 cm−1 for asymmetric stretching of –COO−).47 The FTIR spectra show that –COOH of PMAA in the mixed brush is protonated following immersion in the pH = 3 buffer. As a result of treatment in pH = 8 buffer, the same carboxylic acid functionalities lose their protons. The deprotonation of the polyacid chains was both observed for the homopolymer brush film and in the case of mixed brush layers. As expected for PNIPAM, no spectral change was observed as a result of treatment in buffer at different pH values.
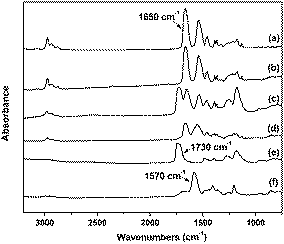 | ||
| Fig. 3 FTIR spectra of (a) PNIPAM homopolymer brush after immersion in pH = 3 buffer, (b) PNIPAM homopolymer brush after immersion in pH = 8 buffer, (c) PNIPAM/PMAA-20 min mixed brush after immersion in pH = 3 buffer, (d) PNIPAM/PMAA-20 min mixed brush after immersion in pH = 8 buffer, (e) PMAA homopolymer brush after immersion in pH = 3 buffer, and (f) PMAA homopolymer brush after immersion in pH = 8 buffer. | ||
The surface morphology of the different polymer brush films following treatment at acidic and basic pH values was subsequently studied by tapping mode AFM (TM-AFM).48 One-component brush samples of PNIPAM and PMAA revealed fairly smooth surfaces following treatment with buffers at different pH (Fig. 4a–f and Table 1). For mixed brushes a sharp morphological transition was observed by immersing the films alternatively in pH = 3 and pH = 8 (Fig. 4g and h). Following immersion in pH = 3 buffer both polymer components were uncharged. The root mean square (RMS) roughness value derived from the corresponding micrograph (Fig. 4g) resulted in a value of 1.80 nm (scan area = 1 × 1 μm2; see Table 1). Charging of the PMAA chains at high pH values and their consequent extension leads to confinement of PNIPAM aggregates close to the substrate surface while the PMAA extends out at the interface. As a result, the RMS roughness values of the mixed brush surface markedly increased to 4.65 nm over a scan area of 1 × 1 μm2 (Fig. 4h). The difference in the roughness values was very significant, thus it can be expected that surface roughness plays a significant role in the contact angle results.
| Contact angle at pH = 3/° | RMS roughness/nm | Contact angle at pH = 8/° | RMS roughness/nm | |
|---|---|---|---|---|
| PNIPAM | 56 ± 2 | 0.37 | 56 ± 2 | 0.33 |
| PMAA | 52 ± 2 | 0.13 | <10 | 0.17 |
| PNIPAM/PMAA-20 min mixed brush | 60 ± 5 | 1.80 | 80 ± 5 (gradually decreasing to 20) | 4.65 |
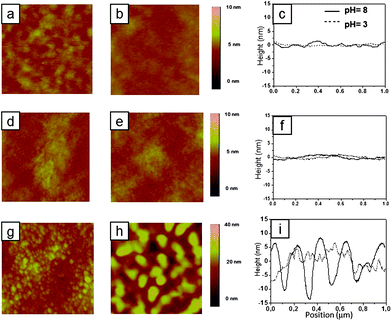 | ||
| Fig. 4 AFM surface height images in air and the representative cross-section of various polymer brush systems following treatments with buffers of different pH (scan area = 1 × 1 μm2). (a) PNIPAM homopolymer brush after immersion in pH = 3 buffer, (b) PNIPAM homopolymer brush after immersion in pH = 8 buffer, and (c) the corresponding cross-sections; (d) PMAA homopolymer brush after immersion in pH = 3 buffer, (e) PMAA homopolymer brush after immersion in pH = 8 buffer, and (f) the corresponding cross-sections; (g) PNIPAM/PMAA-20 min mixed brush after immersion in pH = 3 buffer, (h) PNIPAM/PMAA-20 min mixed brush after immersion in pH = 8 buffer, and (i) the corresponding cross-sections. | ||
Static water contact angle analysis (θ) on the different polymer brush films was subsequently performed and the corresponding results are reported in Table 1. PNIPAM brush surfaces treated with different buffer solutions display similar wettability (contact angle 56°) suggesting no response to pH changes. For PMAA surfaces, a pH change from pH = 3 to pH = 8 induced a shift in contact angle values from 52° for the collapsed state to 10° for the highly hydrophilic swollen state. Similar behavior has already been reported.47 For the mixed brush sample PNIPAM/PMAA-20 min, the contact angle value was quite different compared to single component PMAA and PNIPAM brushes at the corresponding pH values. It is known that the surface roughness can influence the apparent contact angles through many different and subtle ways and contact angles are only affected by the first few nanometres of contacted solids at the air/liquid interface.15,49 At pH = 3, the RMS roughness is 1.80 nm and the contact angle is similar for both PNIPAM and PMAA brushes. However, at pH = 8, the RMS roughness increased to 4.65 nm in the mixed brushes (see Fig. 4). This behavior was paralleled by a contact angle increase to 80 ± 5° (see Table 1) in line with earlier reports. High contact angles were observed even on hydrophilic surfaces due to high surface roughness.50 Since both brushes at pH = 8 are hydrophilic, the high starting contact angle at pH = 8 decreases gradually to 20° in one minute due to the brush reorganization upon water drop deposition.
4. Conclusions
The successful synthesis of binary, mixed PNIPAM/PMAA brushes on gold surfaces at room temperature is described. A simple approach from binary mixed monolayers of initiators by combining two surface-initiated controlled radical polymerization methods, i.e. SI-ATRP and SI-IMP, was employed. The method enables one to achieve height control for both components of the brushes independently in contrast to the previously reported mixed brushes grafted-from silicon and the other grafted-to methods. We found that the mixed brushes undergo reorganization in response to changes in pH, exhibiting reversible changes in swollen/collapsed thicknesses, water contact angles and surface morphology. The stimulus responsive behavior unveiled in this study could find applications in creating functional thin films with reversibly switchable surfaces or adaptive chemical properties.Acknowledgements
We thank the MESA+ institute for Nanotechnology, and the Netherlands Organization for Scientific Research (NWO, TOP Grant 700.56.322, Macromolecular Nanotechnology with Stimulus Responsive Polymers) for financial support.References
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- S. T. Milner, Science, 1991, 251, 905–914 CrossRef CAS.
- E. M. Benetti, S. Zapotoczny and G. J. Vancso, Adv. Mater., 2007, 19, 268–271 CrossRef CAS.
- S. Zapotoczny, E. M. Benetti and G. J. Vancso, J. Mater. Chem., 2007, 17, 3293–3296 RSC.
- M. Navarro, E. M. Benetti, S. Zapotoczny, J. A. Planell and G. J. Vancso, Langmuir, 2008, 24, 10996–11002 CrossRef CAS.
- X. F. Sui, J. Y. Yuan, W. Z. Yuan and M. Zhou, Prog. Chem. (Beijing, China), 2008, 20, 1122–1127 Search PubMed.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- Y. Tsujii, K. Ohno, S. Yamamoto, A. Goto and T. Fukuda, Adv. Polym. Sci., 2006, 197, 1–45 CAS.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- S. G. Boyes, A. M. Granville, M. Baum, B. Akgun, B. K. Mirous and W. J. Brittain, Surf. Sci., 2004, 570, 1–12 CrossRef CAS.
- S. Minko, Polym. Rev., 2006, 46, 397–420 Search PubMed.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H. A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS.
- M. C. LeMieux, S. Peleshanko, K. D. Anderson and V. V. Tsukruk, Langmuir, 2007, 23, 265–273 CrossRef CAS.
- B. Zhao, Langmuir, 2004, 20, 11748–11755 CrossRef CAS.
- B. Zhao, R. T. Haasch and S. MacLaren, J. Am. Chem. Soc., 2004, 126, 6124–6134 CrossRef CAS.
- S. Minko, M. Müller, M. Motornov, M. Nitschke, K. Grundke and M. Stamm, J. Am. Chem. Soc., 2003, 125, 3896–3900 CrossRef CAS.
- P. Uhlmann, L. Ionov, N. Houbenov, M. Nitschke, K. Grundke, M. Motornov, S. Minko and M. Stamm, Prog. Org. Coat., 2006, 55, 168–174 CrossRef CAS.
- T. P. Russell, Science, 2002, 297, 964–967 CrossRef CAS.
- D. Julthongpiput, Y. H. Lin, J. Teng, E. R. Zubarev and V. V. Tsukruk, J. Am. Chem. Soc., 2003, 125, 15912–15921 CrossRef CAS.
- L. Ionov, A. Sidorenko, M. Stamm, S. Minko, B. Zdyrko, V. Klep and I. Luzinov, Macromolecules, 2004, 37, 7421–7423 CrossRef CAS.
- S. Minko, I. Luzinov, V. Luchnikov, M. Müller, S. Patil and M. Stamm, Macromolecules, 2003, 36, 7268–7279 CrossRef CAS.
- N. Houbenov, S. Minko and M. Stamm, Macromolecules, 2003, 36, 5897–5901 CrossRef CAS.
- B. Zhao, Polymer, 2003, 44, 4079–4083 CrossRef CAS.
- S. Santer, A. Kopyshev, H. K. Yang and J. Rühe, Macromolecules, 2006, 39, 3056–3064 CrossRef CAS.
- J. X. Feng, R. T. Haasch and D. J. Dyer, Macromolecules, 2004, 37, 9525–9537 CrossRef CAS.
- E. Bittrich, M. Kuntzsch, K. J. Eichhorn and P. Uhlmann, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1606–1615 CrossRef CAS.
- M. Kaholek, W. K. Lee, J. X. Feng, B. LaMattina, D. J. Dyer and S. Zauscher, Chem. Mater., 2006, 18, 3660–3664 CrossRef CAS.
- S. B. Rahane, J. A. Floyd, A. T. Metters and S. M. Kilbey, Adv. Funct. Mater., 2008, 18, 1232–1240 CrossRef CAS.
- A. J. Parnell, S. J. Martin, C. C. Dang, M. Geoghegan, R. A. L. Jones, C. J. Crook, J. R. Howse and A. J. Ryan, Polymer, 2009, 50, 1005–1014 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
- M. Niwa, M. Date and N. Higashi, Macromolecules, 1996, 29, 3681–3685 CrossRef CAS.
- R. Heeb, R. M. Bielecki, S. Lee and N. D. Spencer, Macromolecules, 2009, 42, 9124–9132 CrossRef CAS.
- R. R. Shah, D. Merreceyes, M. Husemann, I. Rees, N. L. Abbott, C. J. Hawker and J. L. Hedrick, Macromolecules, 2000, 33, 597–605 CrossRef CAS.
- D. M. Jones, A. A. Brown and W. T. S. Huck, Langmuir, 2002, 18, 1265–1269 CrossRef CAS.
- H. Schönherr and H. Ringsdorf, Langmuir, 1996, 12, 3891–3897 CrossRef.
- H. Schönherr, H. Ringsdorf, M. Jaschke, H. J. Butt, E. Bamberg, H. Allinson and S. D. Evans, Langmuir, 1996, 12, 3898–3904 CrossRef.
- E. M. Benetti, E. Reimhult, J. de Bruin, S. Zapotoczny, M. Textor and G. J. Vancso, Macromolecules, 2009, 42, 1640–1647 CrossRef CAS.
- L. H. Li, Y. Zhu, B. Li and C. Y. Gao, Langmuir, 2008, 24, 13632–13639 CrossRef CAS.
- W. Zhang, X. L. Zhu, Z. P. Cheng and J. Zhu, J. Appl. Polym. Sci., 2007, 106, 230–237 CrossRef CAS.
- W. Zhang, X. L. Zhu, J. Zhu and J. Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 32–41 CrossRef CAS.
- R. Nicolay, Y. Kwak and K. Matyjaszewski, Macromolecules, 2008, 41, 4585–4596 CrossRef CAS.
- Y. Kwak, R. Nicolay and K. Matyjaszewski, Macromolecules, 2008, 41, 6602–6604 CrossRef CAS.
- Y. Kwak and K. Matyjaszewski, Macromolecules, 2008, 41, 6627–6635 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS.
- Z. Y. Bao, M. L. Bruening and G. L. Baker, Macromolecules, 2006, 39, 5251–5258 CrossRef CAS.
- A. J. Parnell, S. J. Martin, R. A. L. Jones, C. Vasilev, C. J. Crook and A. J. Ryan, Soft Matter, 2009, 5, 296–299 RSC.
- R. Dong, M. Lindau and C. K. Ober, Langmuir, 2009, 25, 4774–4779 CrossRef CAS.
- X. F. Sui, S. Zapotoczny, E. M. Benetti, P. Schön and G. J. Vancso, J. Mater. Chem., 2010, 20, 4981–4993 RSC.
- C. Dorrer and J. Rühe, Langmuir, 2008, 24, 1959–1964 CrossRef CAS.
- M. Lemieux, D. Usov, S. Minko, M. Stamm, H. Shulha and V. V. Tsukruk, Macromolecules, 2003, 36, 7244–7255 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |