A facile synthesis of branched poly(ethylene glycol) and its heterobifunctional derivatives†
Zhongyu
Li
and
Ying
Chau
*
Department of Chemical and Biomolecular Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong, China. E-mail: kelizy@ust.hk; ying.chau@ust.hk; Fax: +852-2358 0054; Tel: +852-2358 8935
First published on 4th January 2011
Abstract
Allyl-(PEG-OH)2, a dual-branched poly(ethylene glycol) (PEG) with a latent group amenable for modification at the junction, was successfully synthesized using trimethylolpropane allyl ether (TMPAE), a commercially available compound, as the initiator for anionic polymerization. To demonstrate the versatility of this approach, derivatives of the branched PEG were formed using simple modification. The chain ends of PEG were modified to inert methoxy groups and active functional groups. Using an orthogonal reaction procedure, the allyl junction was modified to carboxyl and amino group. The synthesis route was short, quantitative, and easily controlled. No cumbersome purification was needed. The branched PEG and its derivatives were characterized by SEC, 1H and 13C NMR, and MALDI-TOF mass spectroscopy.
Introduction
Poly(ethylene glycol) (PEG), a polymer consisting of ethylene oxide as repeating units, is widely used in biotechnology and medicine due to its excellent safety record in clinical use and its many peculiar properties, including good solubility in a wide range of organic and aqueous media, polymer backbone flexibility, stability in physiological conditions, non-adhesiveness to protein, biocompatibility and the ease of excretion from living organisms.1 The most common applications are found in drug delivery and biosensor preparation, in which biological macromolecules, colloidal carriers, low molecular weight compounds and device surfaces are modified with PEG.1,2In these applications, some advantages have been observed for dual-branched PEG (PEG2) over linear PEG. Nanomaterials, including carbon nanotubes, gold nanoparticles, and gold nanorods, when grafted by branched polyethylene glycol, were found to possess high aqueous solubility and ultra-long circulation half-life upon intravenous injection into mice.3Protein conjugated with a branched PEG has longer in vivo circulation half-life, improved stability against proteolysis, and reduced immunogenicity compared to linear PEG-proteins. This is explained by the greater hydrodynamic volume of branched PEG-proteins, which slows the rate of renal excretion, and the “umbrella-like” structure of the branched PEG around the protein molecule, which provides better shielding.4 The conjugation of interferon-α2b (IFN-α2b) to a branched PEG (PEG2, 40 K) was studied extensively from experiments to clinical phases4d, 5 and is now a product for the treatment of hepatitis C under the trade name PEGASYS.
Until now, almost all branched PEGs have been synthesized by cumbersome methods involving tri-functional linkers. A common method uses lysine to couple two methoxy PEG (mPEG) chains to its alpha and epsilon amino groups and leaves the carboxylic group to be activated for subsequent protein conjugation.4d, 5a, 5b Similarly, 2-(2-aminoethoxy)ethanol6 and tert-butyl protected N,N-bis(2-hydroxyethyl)glycine (bicine)7 have been used as tri-functional linkers for the synthesis of branched PEG. The disadvantages are: 1) multiple and lengthy reactions steps; 2) difficult and expensive purification (using chromatography) for the separation of PEG2 and linear PEG chains; 3) the limitation of the active functional group to be carboxyl; 4) the water susceptibility of the bond between PEG chains and the tri-functional linker (e.g., urethane4d, 6 and ester7 linkage).
Branched PEG synthesized by the polymerization of ethylene oxide (EO) from an initiator containing two hydroxyl groups and one protected or latent group seems to circumvent these problems. Protected glycerol is an example of this kind of initiator. From this initiator, a series of branched PEG derivatives have been synthesized and commercialized by NOF Corporation Ltd. (Tokyo, Japan).8 However, this particular initiator has different reactivity at the alpha and beta hydroxyl positions, making it difficult to control the evenness of PEG chain lengths at the branches.
We report herein a one-pot, inexpensive and high-yielding method to synthesize branched PEG carrying two chains of the statistically same length. Our strategy is to initiate anionic polymerization of ethylene oxide (EO) from trimethylolpropane allyl ether (TMPAE), a commercially available chemical with two uniform alpha hydroxyl groups and one allyl group. After polymerization, the allyl group can be modified to carboxyl or amino group with simple procedures, while the PEG chain ends can be readily functionalized. These dual-branched, heterobiofunctional PEGs will be useful for drug and protein delivery, surface coating, and for preparing block copolymers with functionalized terminals.
Experimental part
Materials
All starting compounds were used as received without additional purification except for those specified. Chemicals were purchased from Aldrich unless otherwise indicated. Tetrahydrofuran (THF) (Merck, 99%) was refluxed over sodium wire and distilled from sodium naphthalenide solution. Trimethylolpropane allyl ether (TMPAE) (Fluka, 98%) and dimethyl sulfoxide (DMSO) (Merck, 98%) were distilled over CaH2 under reduced pressure just before use. Methyl iodide (Riedel deHaer, 99%) was used directly. Ethylene oxide (EO, 99.7%) was purchased from Hong Kong Special Gas Company and used directly. Diphenylmethyl potassium (DPMK) was prepared as described elsewhere.9Methods
1H NMR and 13C NMR spectra were obtained on a DMX 400 MHz spectrometer with tetramethylsilane (TMS) as the internal standard and CDCl3 as the solvent. Size exclusion chromatography (SEC) was performed in 0.1 M NaNO3 at 40 °C with an elution rate of 0.5 mL min−1 on a Waters HPLC system equipped with a G1310A pump and a G1362A refractive index (RI) detector. Ultrahydrogel 250 (Waters) and Ultrahydrogel 1000 (Waters) columns were used in series and calibrated by polyethylene glycol standards (Polymer Source, Inc., Canada). MALDI-TOF MS spectra were recorded using Bruker REFLEX III. α-cyano-4-hydroxycinnamic acid (CHCA) was used as the matrix for the ionization operated in the reflection mode.The synthesis steps of the branched PEG and its derivatives are illustrated in Scheme 1. Details are provided in the following sections.
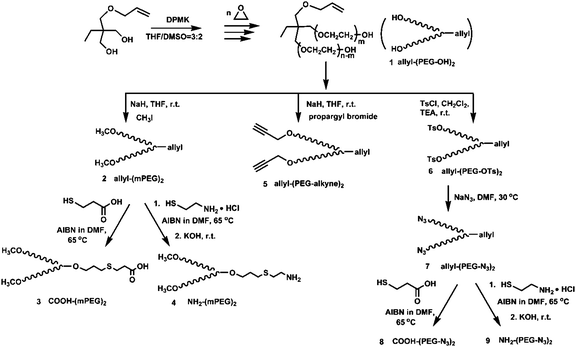 | ||
| Scheme 1 Synthesis of branched PEG (allyl-(PEG-OH)2) and its derivatives | ||
Synthesis of allyl-(PEG-OH)2 (1)
A 150 mL stainless steel kettle was vacuumed at 80 °C for 24 h and cooled to room temperature and then to 0 °C under icy water bath. Anhydrous trimethylolpropane allyl ether (TMPAE) (1.74 g, 0.01mol) was dissolved in 50 mL of mixed solvents of DMSO and THF (v/v: 3/2). A solution of DPMK in THF (6.7 mL, 0.6 M solution) was slowly added. The orange-red color of DPMK was changed to yellow when alkoxide was formed. The homogeneous initiator solution was introduced into the cooled kettle by a syringe, followed with the addition of ethylene oxide (EO). After the solution was stirred at 50 °C for 24 h, polymerization was terminated by adding of a few drops of acidified methanol (0.1 M HCl in methanol). All the solvents were removed by reduced distillation. The crude product was dissolved in CH2Cl2, filtered, and dried over anhydrous MgSO4, then precipitated in diethyl ether. Allyl-(PEG-OH)2 (1) was obtained as a white powder at a reaction yield of 99%. 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2–), 1.38 (q, J = 7.56 Hz, CH3CH2–), 3.25 and 3.28 (s, –C(CH2O–)3, 3.45–3.80 (m, –CH2CH2O– of PEG main chain), 4.02 (d, J = 5.38 Hz, –O–CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.01–5.22 (dd, J = 17.33 Hz and J = 10.25 Hz, –CHCH2), 5.82–5.95 (m, J = 5.13 Hz, –CHCH2); 13C NMR (ppm) (400 MHz, CDCl3): 8.78 (CH3CH2–), 24.5 (CH3CH2–), 43.2 (–C(CH2O–)3, 70.4–71.5 (–CH2CH2O– of PEG main chain), 73.6 (–C(CH2O–)3, 74.2 (–O–CH2–CHCH2), 121.2 (–CHCH2), 132.4 (–CHCH2); SEC: Mn = 5.27 × 103 g mol−1, Mw/Mn = 1.08.
CH2), 5.01–5.22 (dd, J = 17.33 Hz and J = 10.25 Hz, –CHCH2), 5.82–5.95 (m, J = 5.13 Hz, –CHCH2); 13C NMR (ppm) (400 MHz, CDCl3): 8.78 (CH3CH2–), 24.5 (CH3CH2–), 43.2 (–C(CH2O–)3, 70.4–71.5 (–CH2CH2O– of PEG main chain), 73.6 (–C(CH2O–)3, 74.2 (–O–CH2–CHCH2), 121.2 (–CHCH2), 132.4 (–CHCH2); SEC: Mn = 5.27 × 103 g mol−1, Mw/Mn = 1.08.
Synthesis of allyl-(mPEG)2 (2)
Synthesis of allyl-(mPEG)2 was performed according to a method adapted from a previous report.10 One gram (0.38 mmol hydroxyl groups) of allyl-(PEG-OH)2 (Mn = 5.27 × 103 g mol−1) was removed moisture by azeotropic distillation with toluene just before use, then reacted with 0.01 g of NaH (0.42 mmol) in 10 mL of anhydrous tetrahydrofuran (THF) at room temperature under a nitrogen stream for 20 min. This was followed by the addition of 0.10 g of methyl iodide (0.7 mmol) under vigorous stirring at room temperature for 24 h. After neutralization by 0.1 M HCl solution, the solvent was removed by rotary evaporation. The product was dissolved in water and was extracted by dichloromethane (DCM). The organic phase was dried over anhydrous MgSO4. After filtration and concentration, the polymer was precipitated in cold diethyl ether twice to afford a white powder (yield = 95%). 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2–), 1.38 (q, J = 7.56 Hz, CH3CH2–), 3.25 and 3.28 (s, –C(CH2O–)3, 3.38 (s, CH3O–), 3.45–3.80 (m, –CH2CH2O– of PEG main chain), 4.02 (d, J = 5.38 Hz, –O–CH2–CHCH2), 5.01–5.22 (dd, J = 17.33 Hz and J = 10.25 Hz, –CHCH2), 5.82–5.95 (m, J = 5.13 Hz, –CHCH2); SEC: Mn = 5.28 × 103 g mol−1, Mw/Mn = 1.08.
Synthesis of COOH-(mPEG)2 (3)
The carboxylation reaction of the allyl terminus of polymer allyl-(mPEG)2 was conducted by the radical addition reaction using 3-mercaptopropionic acid.10,11Allyl-(mPEG)2 (1 g, 0.2 mmol) with moisture removed by azeotropic distillation with toluene just before use, was mixed with a solution containing 424 mg of 3-mercaptopropionic acid (4 mmol, 20 equivalent) and 36.0 mg of azobisisobutyronitrile (AIBN) (0.1 mmol, 1 equivalent) in 5 mL anhydrous dimethylformamide (DMF). The reaction mixture was stirred at 65 °C for 24 h under nitrogen atmosphere. The polymer was precipitated twice in a large excess of diethyl ether. The polymer COOH-(mPEG)2 was obtained as a white power (924 mg, yield = 84%). 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2–), 1.38 (q, J = 7.56 Hz, CH3CH2–), 1.76 (p, J = 6.71 Hz, –OCH2CH2CH2S–), 2.56 (t, J = 7.08 Hz, –CH2CH2–S–CH2– and t, J = 7.32 Hz, –S–CH2CH2COOH), 2.96 (t, J = 7.32 Hz and J = 7.08 Hz, –CH2COOH), 3.25 and 3.28 (s, –C(CH2O–)3, 3.38 (s, CH3O–), 3.45–3.80 (m, –CH2CH2O– of PEG main chain and –OCH2CH2CH2S–); SEC: Mn = 5.28 × 103 g mol−1, Mw/Mn = 1.08.Synthesis of NH2-(mPEG)2 (4)
In a typical reaction, 400 mg of allyl-(mPEG)2 (0.077 mmol) in 10 mL of anhydrous DMF was reacted with 175 mg of 2-aminoethanethiol hydrochloride (1.54 mmol, 20 equiv.) in the presence of 12.6 mg of AIBN (0.077 mmol, 1 equiv). The reaction mixture was stirred at 65 °C for 24 h under nitrogen atmosphere. After the reaction, the solution was precipitated in diethyl ether twice. The resulting white product was dissolved in methanol, and 4.3 mg (0.077 mmol, 1 equiv) of potassium hydroxide dissolved in water was added. The mixture was stirred for approximately 4 h. Then, methanol was partially evaporated and diluted with water (30 mL), and extracted by dichloromethane (3 × 50 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated. The polymer was reprecipitated from an excess volume of ether twice to afford a white powder (412 mg, yield = 79.5%). 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2–), 1.38 (q, J = 7.56 Hz, CH3CH2–), 1.76 (p, J = 6.71 Hz, –OCH2CH2CH2S–), 2.41 (t, J = 7.57 Hz, –CH2–S–CH2–), 2.66 (t, J = 7.57 Hz, –CH2–S–CH2–), 2.94 (t, J = 7.08 Hz, –CH2CH2NH2), 3.25 and 3.28 (s, –C(CH2O–)3, 3.38 (s, CH3O–), 3.45–3.80 (m, –CH2CH2O– of PEG main chain and –OCH2CH2CH2S–); SEC: Mn = 5.28 × 103 g mol−1, Mw/Mn = 1.08.Synthesis of allyl-(PEG-alkyne)2 (5)
Synthesis of allyl-(PEG-alkyne)2 was performed using a method adapted from a previous report.12 One gram (0.38 mmol hydroxyl groups)of allyl-(PEG-OH)2 (Mn = 5.27 × 103 g mol−1), with moisture removed by azeotropic distillation with toluene just before use was mixed with 0.12 g NaH (0.5 mmol) in 10 mL anhydrous THF under nitrogen atmosphere at room temperature for 1 h, then 0.060 g propargyl bromide (0.5 mmol) was added at room temperature for 24 h. After neutralization by hydrogen chloride solution, the solvent was removed by rotary evaporation. The crude product was dissolved in water and extracted by DCM (3 × 50 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated. The polymer was reprecipitated from an excess volume of diethyl ether twice to afford a white powder (835 mg, yield = 83%). 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2–), 1.38 (q, J = 7.56 Hz, CH3CH2–), 2.40 (s, J = 2.19 Hz, –CCH), 3.25 and 3.28 (s, –C(CH2O–)3, 3.45–3.80 (m, –CH2CH2O– of PEG main chain), 4.02 (d, J = 5.38 Hz, –O–CH2–CHCH2), 4.17 (s, J = 2.44 Hz, –CH2–CCH), 5.01–5.22 (dd, J = 17.33 Hz and J = 10.25 Hz, –CHCH2), 5.82–5.95 (m, J = 5.13 Hz, –CHCH2); SEC: Mn = 5.28 × 103 g mol−1, Mw/Mn = 1.08.
Synthesis of allyl-(PEG-N3)2 (7)
The azide end group was introduced by the tosylation of the hydroxyl terminus and the subsequent substitution with sodium azide, in accordance with a previously reported method.12 One gram (0.38 mmol hydroxyl groups) of allyl-(PEG-OH)2 (Mn = 5.27 × 103 g mol−1) with moisture removed by azeotropic distillation with toluene just before use was dissolved in anhydrous THF (10 mL), followed by the addition of triethylamine (40 mg, 0.4 mmol). The mixture was then added to a solution of p-toluenesulfonyl chloride (57 mg, 0.5 mmol) in THF (8 mL) under nitrogen atmosphere, and stirred overnight at room temperature. After the reaction, THF was partially evaporated under reduced pressure. The residue was dissolved in water (20 mL) and extracted with dichloromethane (3 × 50 mL). The organic layers were combined and dried over anhydrous MgSO4. After filtration and concentration, the polymer was recovered by precipitation into diethyl ether and dried in vacuo, yielding a white powder (6) (yield = 87%). Allyl-(PEG-OTs)2: 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2–), 1.38 (q, J = 7.56 Hz, CH3CH2–), 2.45 (s, CH3–C6H4–), 3.25 and 3.28 (s, –C(CH2O–)3, 3.45–3.80 (m, –CH2CH2O– of PEG main chain), 4.02 (d, J = 5.38 Hz, –O–CH2–CHCH2), 5.01–5.22 (dd, J = 17.33 Hz and J = 10.25 Hz, –CHCH2), 5.82–5.95 (m, J = 5.13 Hz, –CHCH2); 7.35 (d, J = 7.82 Hz, two CH in phenyl ring close to –CH3), 7.79 (d, J = 7.81 Hz, other two CH in phenyl ring).
This tosylated polymer (6) (500 mg, 0.1 mmol) was dissolved in anhydrous DMF (12 mL), followed by sodium azide (1.3 mg, 2 mmol) addition, and was stirred for 2 days at 30 °C. DCM (100 mL) was then added, and the reaction mixture was washed five times with water and brine. The organic layer was dried over anhydrous MgSO4, filtered, concentrated, and then precipitated in diethyl ether. Allyl-(PEG-N3)2 (7) was obtained as a white powder (447 mg, yield = 92%). 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2–), 1.38 (q, J = 7.56 Hz, CH3CH2–), 1.78 (m, J = 7.28 Hz, –CH2CH2N3); 3.25 and 3.28 (s, –C(CH2O–)3, 3.45–3.80 (m, –CH2CH2O– of PEG main chain and –CH2CH2N3), 4.02 (d, J = 5.38 Hz, –O–CH2–CHCH2), 5.01–5.22 (dd, J = 17.33 Hz and J = 10.25 Hz, –CHCH2), 5.82–5.95 (m, J = 5.13 Hz, –CHCH2); 13C NMR (ppm) (400 MHz, CDCl3): 8.78 (CH3CH2–), 24.5 (CH3CH2–), 43.2 (–C(CH2O–)3, 50.8 (–CH2CH2N3), 69.5 (–CH2CH2N3), 70.4–71.5 (–CH2CH2O– of PEG main chain), 73.6 (–C(CH2O–)3, 74.2 (–O–CH2–CHCH2), 117.5 (–CHCH2), 132.4 (–CHCH2); SEC: Mn = 5.28 × 103 g mol−1, Mw/Mn = 1.08.
Synthesis of COOH-(PEG-N3)2 (8) and NH2-(PEG-N3)2 (9)
The synthesis of COOH-(PEG-N3)2 and NH2-(PEG-N3)2 are similar to the synthesis of COOH-(mPEG)2 and NH2-(mPEG)2, respectively. Instead of using allyl-(mPEG)2 (2) as the polymer for modification, (allyl-(PEG-N3)2 (7) was used.COOH-(PEG-N3)2 (8) was obtained as a white powder with a reaction yield of 94%. 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2–), 1.38 (q, J = 7.56 Hz, CH3CH2–), 1.76 (p, J = 6.71 Hz, –OCH2CH2CH2S–), 1.82 (t, J = 7.28 Hz, –CH2CH2N3), 2.56 (t, J = 7.08 Hz, –CH2CH2–S–CH2– and t, J = 7.32 Hz, –S–CH2CH2COOH), 2.96 (t, J = 7.32 Hz and J = 7.08 Hz,, –CH2COOH), 3.25 and 3.28 (s, –C(CH2O–)3, 3.38 (s, CH3O–), 3.45–3.80 (m, –CH2CH2O– of PEG main chain, –OCH2CH2CH2S– and –CH2CH2N3); 13C NMR (ppm) (400 MHz, CDCl3): 8.78 (CH3CH2–), 24.5 (CH3CH2–), 27.1 (–SCH2CH2COOH), 34.8 (–SCH2CH2COOH), 40.2 (–C(CH2O–)3, 50.8 (–CH2CH2N3), 69.5 (–CH2CH2N3), 70.4–71.5 (–CH2CH2O– of PEG main chain), 73.6 (–C(CH2O–)3, 173.4 (–SCH2CH2COOH); SEC: Mn = 5.29 × 103 g mol−1, Mw/Mn = 1.08.
NH2–(PEG-N3)2 (9) was obtained as a white powder with a reaction yield of 91%. 1H NMR (ppm) (400 MHz, CDCl3): 0.84 (t, J = 7.57 Hz, CH3CH2-), 1.38 (q, J = 7.56 Hz, CH3CH2–), 1.76 (p, J = 6.71 Hz, –OCH2CH2CH2S–), 1.82 (t, J = 7.28 Hz, –CH2CH2N3), 2.41 (t, J = 7.57 Hz, –CH2–S–CH2–), 2.66 (t, J = 7.57 Hz, –CH2–S–CH2–), 2.94 (t, J = 7.08 Hz, –CH2CH2NH2), 3.25 and 3.28 (s, –C(CH2O–)3, 3.38 (s, CH3O–), 3.45–3.80 (m, –CH2CH2O– of PEG main chain, –OCH2CH2CH2S– and –CH2CH2N3); 13C NMR (ppm) (400 MHz, CDCl3): 8.78 (CH3CH2–), 24.5 (CH3CH2–), 28.5 (–SCH2CH2NH2), 40.3 (–SCH2CH2NH2), 43.2 (–C(CH2O–)3, 50.8 (–CH2CH2N3), 69.5 (–CH2CH2N3), 70.4–71.5 (–CH2CH2O– of PEG main chain), 73.6 (–C(CH2O–)3; SEC: Mn = 5.30 × 103 g mol−1, Mw/Mn = 1.08.
Results and discussion
Much attention has been given to improve the synthesis of branched PEG.4d, 6–8 We are motivated to develop a novel procedure to overcome the drawbacks of conventional coupling methods, which require multiple synthesis steps and difficult purification. The choice of trimethylolpropane allyl ether (TMPAE) as the initiator is inspired by the facile synthesis of heterobifunctional PEG initiated from allyl alcoholate.10,11 TMPAE allows the synthesis of branched PEG with equal chain lengths viaanionic polymerization of EO because the allyl group is inert for the polymerization and does not affect the reactivity of the two pendent alpha hydroxyl groups. In addition, the allyl group is versatile and enables easy chemical modifications such as addition reactions. The polymerization was carried out in mixed solvent (THF/DMSO = 3/2) as reported by us before9 and the reaction time was dependent on the pre-designed number average molecular weight (Mn). It was found that the polymerization of branched PEG of Mn = 5k and 20k required 24 h and 96 h respectively. After precipitation into ether, the product was obtained as a white powder. Results of polymerization using TMPAE as an initiator under different conditions are summarized in Table 1. The molecular weight of the polymers (allyl-(PEG-OH)2) (1) determined from SEC was close to that calculated by the initial monomer/initiator ratio, supporting that the polymerization was complete and without detrimental side reactions. The resulting polymers are unimodal with narrow polydispersity (PDI) (Mw/Mn). The value of the Mn could be controlled by the initial monomer/initiator ratio while retaining a narrow polydispersity due to the nature of anionic polymerization.| [EO]0/[TMPAE]0 | 10−3 × Mnb (g mol−1) | Polydispersity Mw/Mng | |||||
|---|---|---|---|---|---|---|---|
| Calcdc | NMR d | SEC e | MS f | SEC g | MS h | ||
| a Reaction time of 1 and 2 are 24 h and 96 h respectively, and the yields of product 1 and 2 are 99% and 99% respectively. b M n denotes number average molecular weight. c Determined from the following equation: Mn(calcd) = Mw(EO)[EO]0/[TMPAE]0 + Mw(TMPAE) = 44.05 [EO]0/[TMPAE]0 + 174.23 (3). d The number average molecular weight (Mn) of the polymers was determined by 1H NMR spectrum on the basis of end group analysis using the eqn (1). e Determined by SEC. f Determined by MALDI-TOF mass spectroscopy. g Polydispersity Index (PDI) = Mw/Mn. Mw denotes weight average molecular weight. h The PDI according to MALDI-TOF MS. | |||||||
| 1 a | 110 | 5.01 | 5.24 | 5.27 | 5.36 | 1.08 | 1.03 |
| 2 a | 450 | 20.02 | 20.64 | 20.85 | ND | 1.09 | ND |
The structure of allyl-(PEG-OH)2 was confirmed by 1H NMR and 13C NMR. In the 1H NMR spectrum (Fig. 1A), the signals of the protons of the allyl group are detected at δ 4.02 ppm (d, –O–CH2–CHCH2), 5.01–5.22 ppm (dd, –CHCH2), 5.82–5.95 ppm (m, –CHCH2), respectively. The number average molecular weight (Mn) of the polymers was determined by 1H NMR spectrum on the basis of end group analysis using the following equation:
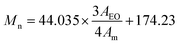 | (1) |
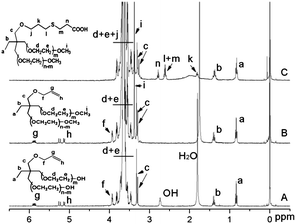 | ||
| Fig. 1 1H NMR spectra of allyl-(PEG-OH)2 (A), allyl-(mPEG)2 (B) and COOH-(mPEG)2 (C) respectively, (CDCl3 at 20 °C). | ||
Allyl-(PEG-OH)2 of pre-designed Mn, at 5k was further characterized by MALDI-TOF MS spectroscopy (Fig. 2). The polymer was confirmed to be unimodal and narrowly distributed (Mw/Mn = 1.03). Presence of side reaction product was not indicated. The molecular weight found using MALDI-TOF was 5.36k, was in good agreement with the SEC and NMR results. The major series of the molecular masses of the product is expressed in the following equation:
| Mw(MALDI-TOF) = 44.035n(EO) + 174.23(TMPAE) + 22.99(sodium) | (2) |
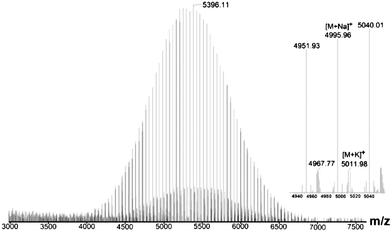 | ||
| Fig. 2 MALDI-TOF Mass spectrum of allyl-(PEG-OH)2. | ||
The polydispersities (PDIs) of allyl-(PEG-OH)2 characterized by MOLDI-Tof MS are lower than those characterized by SEC (Table 1). This phenomenon has been discussed in previous report13 and the authors considered that the reason may be a small amount of SEC axial dispersion.
From (1), we capped the terminal hydroxyl groups with methyl iodine to prepare allyl-(mPEG)2 (2). Fig. 1A and Fig. 1B show the 1H NMR spectrum of PEG before and after methylation. The new peak at δ = 3.38 ppm was assigned to methoxy end group in Fig. 1B. The peak area ratio of terminal methoxy group (δ = 3.38 ppm) to methyl group (δ = 0.84 ppm) was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, showing that the hydroxyl groups were completely methylated. The SEC result indicated the modification maintained the unimodality and narrow polydispersity of the polymer.
:1, showing that the hydroxyl groups were completely methylated. The SEC result indicated the modification maintained the unimodality and narrow polydispersity of the polymer.
The allyl group is not an active group for bioconjugation, but it can be modified to carboxyl, amino or hydroxyl group easily by thiol-ene “click” reaction,14 a hydrothiolation of a double bond with chemicals containing thiol group, such as 3-mercaptopropionic acid, 2-aminoethanethiol hydrochloride and 2-mercaptoethanol. To activate the branched allyl-(mPEG)2, the radical addition of 3-mercaptopropionic acid and 2-aminoethanethiol hydrochloride to the allyl middle group of the polymer was performed. Taking COOH-(mPEG)2 (3) as an example, the 1H NMR spectrum (Fig. 1C) of the polymer revealed the complete disappearance of the signals assigned to the allyl protons at 4.02 ppm (d, –O–CH2–CHCH2), 5.01–5.22 ppm (dd, –CHCH2), and 5.82–5.95 ppm (m, –CHCH2). Concomitantly, new signals were clearly observed at 1.76 ppm (m, –OCH2CH2CH2S–), 2.56 ppm (m, –CH2–S–CH2–), and 2.96 ppm (m, –CH2COOH), corresponding to the resulting structure via the addition of 3-mercapopropionic acid to allyl group. Synthesis of NH2-(mPEG)2 was also carried out (Scheme 1). The 1H NMR spectrum of this polymer also revealed that the allyl groups were completely transformed into amino groups based on the complete disappearance of the signals assigned to the allyl protons and the appearance of the signals at 1.76 ppm (m, –OCH2CH2CH2S–), 2.41 ppm (m, –CH2–S–CH2–), 2.66 ppm (m, –CH2–S–CH2–) and 2.94 ppm (m, –CH2CH2NH2) (Spectrum not shown). Thus two functionalized dual-branched mPEGs (COOH–(mPEG)2 and NH2–(mPEG)2) have been successfully synthesized. They are particularly suitable for drug, peptide, or protein pegylation.
Allyl-(PEG-OH)2 is a starting material for preparing heterobifunctional branched PEG derivatives containing two uniform end groups and another active group at the mid-junction. Five heterobifunctional dual-branched PEGs have been successfully synthesized: allyl-(PEG-alkyne)2 (5), allyl-(PEG-OTs)2 (6), allyl-(PEG-N3)2 (7), COOH-(PEG-N3)2 (8), NH2-(PEG-N3)2 (9). From the 1H NMR spectrum (Fig. 3) of allyl-(PEG-alkyne)2, new signals at 2.40 ppm (s, –CCH) and 4.17 ppm (s, –CH2–CCH), along with the 2:1 molar ratio of alkyne group (δ = 2.40 and 4.17 ppm) to methoxy group (δ = 0.84 ppm), showed that the hydroxyl groups were completely changed to alkyne group. Alkyyne group is amenable for “click chemistry”, a highly specific cycloaddition that can take place in mild aqueous conditions.15
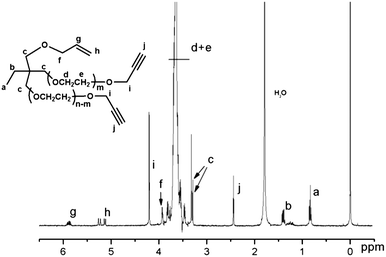 | ||
| Fig. 3 1H NMR spectrum of allyl-(PEG-alkyne)2 (CDCl3 at 20 °C). | ||
Two other clickable heterobifunctional branched PEG, COOH-(PEG-N3)2 (8) and NH2-(PEG-N3)2 (9), were successfully synthesized. Aside from 1H NMR (with identical spectra of (3) and (8), and of (4) and (9)), 13C NMR gave strong evidence about the synthesis of these derivatives. As Fig. 4 shows, the corresponding carbon signals at 121.2 ppm (–CHCH2) and 132.4 ppm (–CHCH2) completely disappeared and new peaks at 27.1 ppm (–SCH2CH2COOH), 34.8 ppm (–SCH2CH2COOH), and 173.4 ppm (–SCH2CH2COOH) were observed, indicating the conversion of allyl group to carboxyl group. Meanwhile, no side reaction of the azido group such as radical scavenging was detected. These results indicated the successful preparation of heterobifunctional, dual-branched PEG possessing carboxyl group on the chain middle and azido groups on the two chain ends. These heterobifunctional dual-branched PEGs may find applications in combination drug delivery16 or targeting drug delivery,17 and biosensors18 and surface modification.19 It should be pointed out that in addition to those derivatives demonstrated in this report, a large number of heterobifunctional dual-branched PEGs can be designed and synthesized due to the versatility of the hydroxyl groups present on allyl-(PEG-OH)22,12
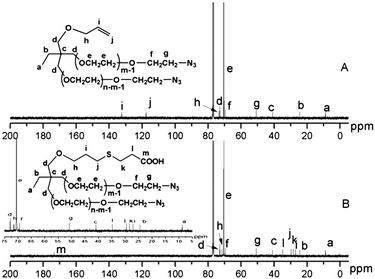 | ||
| Fig. 4 13C NMR spectra of allyl-(PEG-N3)2 (A) and COOH-(PEG-N3)2 (B), (CDCl3 at 20 °C). | ||
Conclusion
In conclusion, a new and well-defined dual-branched PEG, allyl-(PEG-OH)2, was successfully synthesized with a commercial chemical TMPAE as the initiator for the anionic polymerization of ethylene oxide. The polymer is modifiable for a number of derivatives. The preparation of branched PEGs with methoxy terminals, having carboxyl or amino group in the mid-junction, and heterobifunctional branched PEGs was demonstrated. The synthesis and purification procedures were short and simple. These dual-branched mPEG and heterofunctional dual-branched PEG can find applications in drug and protein delivery and surface modification of biosensors.Acknowledgements
The authors gratefully acknowledge financial support from the Hong Kong Research Grant Council (General Research Fund 600207).Notes and references
- (a) J. M. Harris, Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications, Plenum Publishing corporation, New York, 1992 Search PubMed; (b) J. M. Harris and S. Zalipsky, Poly(ethylene glycol) Chemistry and Biological Applications, American Chemical Society, Washington. 1997 Search PubMed.
- (a) J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS; (b) M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS; (c) F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS; (d) F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS; (e) F. M. Veronese and G. Pasut, Adv. Drug Delivery Rev., 2009, 61, 1177–1188 CrossRef CAS.
- G. Prencipe, S. M. Tabakman, K. Welsher, Z. Liu, A. P. Goodwin, L. Zhang, J. Henry and H. J. Dai, J. Am. Chem. Soc., 2008, 131, 4783–4787.
- (a) C. J. Fee, Biotechnol. Bioeng., 2007, 98, 725–731 CrossRef CAS; (b) F. M. Veronese, P. Caliceti and O. Schiavon, J. Bioact. Compat. Polym., 1997, 12, 196–207 CAS; (c) F. M. Veronese, C. Monfardini, P. Caliceti, O. Schiavon, M. D. Scrawen and D. Beer, J. Controlled Release, 1996, 40, 199–209 CrossRef CAS; (d) C. Monfardini, O. Schiavon, P. Caliceti, M. Morpurgo, J. M. Harris and F. M. Veronese, Bioconjugate Chem., 1995, 6, 62–69 CrossRef CAS.
- (a) P. Bailon, A. Palleroni, C. A. Schaffer, C. L. Spence, W. J. Fung, J. E. Porter, G. K. Ehrlich, W. Pan, Z. X. Xu, M. W. Modi, A. Farid and W. Berthold, Bioconjugate Chem., 2001, 12, 195–202 CrossRef CAS; (b) K. R. Reddy, M. W. Modi and S. Pedder, Adv. Drug Delivery Rev., 2002, 54, 571–586 CrossRef; (c) A. Kozlowski, S. A. Charles and J. M. Harris, BioDrugs, 2001, 15, 419–429 CrossRef CAS; (d) K. R. Reddy, T. L. Wright, P. J. Pockros, M. Shiffman, G. Everson, R. Reindollar, M. W. Fried, P. P. Purdum, D. Jensen, C. Smith, W. M. Lee, T. D. Boyer, A. Lin, S. Pedder and J. DePamphilis, Hepatology, 2001, 33, 433–438 CrossRef CAS; (e) R. J. Motzer, A. Rakhit, J. Thompson, H. Gurney, P. Selby, R. Figlin, S. Negrier, S. Ernst, M. Siebels, M. Ginsberg, K. Rittweger and L. Hooftman, Ann. Oncol., 2002, 13, 1799–1805 CrossRef CAS; (f) S. Zeuzem, S. V. Feinman, J. Rasenack, E. J. Heathcote, M. Y. Lai, E. Gane, J. O'Grady, J. Reichen, M. Diago, A. Lin, J. Hoffman and M. J. Brunda, N. Engl. J. Med., 2000, 343, 1666–1672 CrossRef CAS; (g) E. J. Heathcote, M. L. Shiffman, W. G. E. Cooksley, G. M. Dusheiko, S. S. Lee, L. Balart, R. Reindollar, R. K. Reddy, T. L. Wright, A. Lin, J. Hoffman and J. De Pamphilis, N. Engl. J. Med., 2000, 343, 1673–1680 CrossRef CAS; (h) S. J. Keam and R. S. Cvetkovic, Drugs, 2008, 68, 1273–1319 CrossRef CAS; (i) S. J. Keam and R. S. Cvetkovic, BioDrugs, 2009, 23, 63–68 Search PubMed.
- A. Martinez, A. Pendri, J. Xia and R. B. Greenwald, Macromol. Chem. Phys., 1997, 198, 2489–2498 CrossRef CAS.
- H. Zhao, K. Yang, A. Martinez, A. Basu, R. Chintala, H. C. Liu, A. Janjua, M. L. Wang and D. Filpula, Bioconjugate Chem., 2006, 17, 341–351 CrossRef CAS.
- (a) NOF Corp., M. Kouzou and K. Yoshiyuki, 1999-08-24; (b) http://www.nof.co.jp/business/dds/apeg/english/con02/con02-2.html-2010-10-14.
- Z. Y. Li, P. P. Li and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4361–4371 CrossRef CAS.
- S. Cammas, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 1995, 6, 226–230 CrossRef CAS.
- S. Hiki and K. Kataoka, Bioconjugate Chem., 2007, 18, 2191–2196 CrossRef CAS.
- Z. Y. Li and Y. Chau, Bioconjugate Chem., 2009, 20, 780–789 CrossRef CAS.
- T. H. Mourey, A. J. Hoteling, S. T. Balke and K. G. Owens, J. Appl. Polym. Sci., 2005, 97, 627–639 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- J. F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958–970 CrossRef CAS.
- A. Warnecke and F. Kratz, Bioconjugate Chem., 2003, 14, 377–387 CrossRef CAS.
- G. Pasut, F. Canal, L. D. Via, S. Arpicco, F. A. Veronese and O. Schiavon, J. Controlled Release, 2008, 127, 239–248 CrossRef CAS.
- R. Schlapak, P. Pammer, D. Armitage, R. Zhu, P. Hinterdorfer, M. Vaupel, T. Fruhwirth and S. Howorka, Langmuir, 2006, 22, 277–285 CrossRef CAS.
- A. K. Pannier, J. A. Wieland and L. D. Shea, Acta Biomater., 2008, 4, 26–39 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The SEC trace of allyl-(PEG-OH)2. See DOI: 10.1039/c0py00339e |
| This journal is © The Royal Society of Chemistry 2011 |