New thioxanthone and xanthone photoinitiators based on silyl radical chemistry†
Jacques
Lalevée
*a,
Nicolas
Blanchard
b,
Mohamad Ali
Tehfe
a,
Cédric
Fries
b,
Fabrice
Morlet-Savary
a,
Didier
Gigmes
c and
Jean Pierre
Fouassier
a
aDepartment of Photochemistry, CNRS, University of Haute Alsace, ENSCMu, 3 rue Alfred Werner, 68093, Mulhouse cedex, France
bDepartment of Organic and Bioorganic Chemistry, CNRS, University of Haute Alsace, ENSCMu, 3 rue Alfred Werner, 68093, Mulhouse cedex, France
cUMR 6264 Laboratoire Chimie Provence, Université de Provence, Avenue Escadrille Normandie-Niemen, Case 542, 13397, Marseille cedex 20, France. E-mail: j.lalevee@uha.fr
First published on 28th February 2011
Abstract
The ability of a new efficient photoinitiator (TX–Si), based on the well-known thioxanthonechromophore linked to a disilylacetylene moiety and exhibiting a red-shifted absorption, to initiate both free radical polymerization and cationic polymerization is checked. A comparison with the parent compound (isopropylthioxanthone) is provided. High rates of polymerization and high final conversions are obtained. The migration of TX–Si out of the polymer film is noticeably reduced (3-fold factor) compared to that observed with ITX. A similar modification of the xanthonechromophore (XT–Si) also leads to an efficient cleavable photoinitiator and opens a door for the design of a new class of potentially interesting compounds for UV curing applications. As characterized by ESR spin trapping, TX–Si and XT–Si generate silyl radicals under light irradiation evidencing a Type I photoinitiator character. The analysis of the excited state processes through laser flash photolysis and molecular orbital calculations allows explaining the observed photochemical behavior.
Introduction
The structural modification of a photoinitiator (PI) skeleton for getting new properties (better absorption, better reactivity, better compatibility, less extractability,…) has continuously received huge attention (see e.g. in ref. 1). Among a lot of PIs, thioxanthone TX was certainly one of the most investigated chromophores. TX derivatives behave as Type II PI (they need the presence of a co-initiator e.g. usually an amine). The 2-isopropyl thioxanthone (ITX) is very popular in industry but its migration and the subsequent film yellowing are sometimes a drawback. Moreover, oxygen inhibition that occurs in free radical polymerization (FRP) and free radical promoted cationic polymerization (FRPCP) is a serious problem in very thin and low viscosity coatings or in UV inkjet printing.The substitution at the 2-, 3,3′- or 1,4- position of TX,2 the incorporation of a H-donor group (such as a thiol, a thioether, a carboxylic acid) to get a one-component Type II PI system (see ref. 3 and references therein), the introduction of an anthracene or a fluorene moiety on the TX backbone (see ref. 3 and references therein) and the synthesis of TX based macro-PI have recently received much attention.4 On the opposite, the structurally related xanthone (XT) has been little reported as PI. All these derivatives still operate, however, as Type II PI (except the thioxanthone–anthracene compound that directly leads, under air, to the generation of radicals3a).
In the present paper, we mainly propose the design of a new TX derivative and show the potential of a new XT derivative, both compounds having to be able to (i) behave as a cleavable Type I PI in order to avoid the harmful character of amines and reduce the migration, (ii) exhibit a better light absorption in the near UV-visible region, and (iii) keep a high efficiency for the initiation of free radical photopolymerization (FRP) and free radical promoted cationic photopolymerization (FRPCP) under air. According to our works using the silyl radical chemistry to design various photoinitiating systems less sensitive to the oxygen inhibition,5 we select here the TX–Si and XT–Si compounds shown in Scheme 1 as starting model derivatives that should meet the desired properties. FTIR spectroscopy, ESR spin trapping, laser flash photolysis and quantum molecular calculations will allow to outline the efficiency and the reactivity of this promising kind of PI.
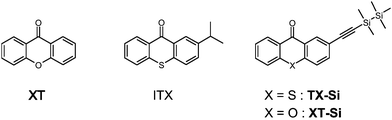 | ||
| Scheme 1 | ||
Experimental section and computational procedure
Samples
To a solution of 2-bromo-thioxanthen-9-one (1 g, 3.43 mmol), copper iodide (5 mol%, 0.033 g), tetrakis(tristriphenylphosphine)palladium (5 mol%, 0.198 g) and triethylamine (6 mL, 42.7 mmol) in dry DMF (20 mL) was added (pentamethyldisilyl)acetylene (0.64 mL, 3.25 mmol) in dry DMF (4 mL) dropwise over 30 min at room temperature.6b The reaction mixture was stirred overnight, diluted with water and extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate, filtered and concentrated under vacuum. The solid residue was adsorbed on silica gel and purified by flash chromatography (cyclohexane/ethyl acetate = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) to give 0.46 g (37%) of 2-((1,1,2,2,2-pentamethyldisilyl)ethynyl)-9H-thioxanthen-9-one as a bright yellow powder, MP = 85–87 °C. 1H NMR (CDCl3; 300 MHz) δ 8.68 (d, J = 1.7 Hz, 1H); 8.61 (dd, J = 8.1 and 1.2 Hz, 1H), 7.63 (dt, J = 8.3 and 1.9 Hz, 2H); 7.56 (m, 1H); 7.52–7.47 (2H). 13C NMR (CDCl3; 75 MHz) δ 179.2; 137.0; 136.8; 134.8; 133.2; 132.4; 129.9; 129.0 (2C); 126.5; 126.0; 125.9; 121.7; 105.7; 95.2; −2.5; −3.1 (ESI†).
:2) to give 0.46 g (37%) of 2-((1,1,2,2,2-pentamethyldisilyl)ethynyl)-9H-thioxanthen-9-one as a bright yellow powder, MP = 85–87 °C. 1H NMR (CDCl3; 300 MHz) δ 8.68 (d, J = 1.7 Hz, 1H); 8.61 (dd, J = 8.1 and 1.2 Hz, 1H), 7.63 (dt, J = 8.3 and 1.9 Hz, 2H); 7.56 (m, 1H); 7.52–7.47 (2H). 13C NMR (CDCl3; 75 MHz) δ 179.2; 137.0; 136.8; 134.8; 133.2; 132.4; 129.9; 129.0 (2C); 126.5; 126.0; 125.9; 121.7; 105.7; 95.2; −2.5; −3.1 (ESI†).
Following the procedure of Larock and Zhao,6c to a solution of 5-bromo-2-hydroxy-benzoic acid methyl ester (0.70 g, 3.0 mmol) in dry THF (60 mL) was added cesium fluoride (1.85 g, 12.2 mmol) and 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (1 g, 3.35 mmol). The reaction mixture was stirred at 65 °C for 24 h, cooled to room temperature and diluted with water. The solution was extracted with diethyl ether and the combined organic phases were dried over magnesium sulfate, filtered and concentrated under vacuum. The residue was purified by flash chromatography on silica gel (cyclohexane/ethyl acetate = 95:5) to give 455 mg (55%) of 2-bromo-9H-xanthen-9-one as a white solid, MP = 147–149 °C (Lit. [6d] 149.3–150.5 °C). 1H NMR (CDCl3; 300 MHz) δ 8.46 (d, J = 2.6 Hz, 1H); 8.33 (dd, J = 7.9 and 1.7 Hz, 1H); 7.82–7.73 (2H); 7.50 (d, J = 8.6 Hz, 1H); 7.44–7.38 (2H). These spectroscopic data are in agreement with those reported in the literature.6c
To a solution of 2-bromo-9H-xanthen-9-one (0.3 g, 1.1 mmol), copper iodide (5 mol%, 10 mg), tetrakis(tristriphenylphosphine)palladium (5 mol%, 63 mg) and triethylamine (2.1 mL,15 mmol) in dry DMF (10 mL) was added (pentamethyldisilyl)acetylene (0.15 mL, 1.1 mmol) in dry DMF (5 mL) dropwise over 30 min at room temperature.6b The reaction mixture was stirred overnight, diluted with water and extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate, filtered and concentrated under vacuum. The solid residue was adsorbed on silica gel and purified by flash chromatography (cyclohexane/ethyl acetate = 100:2) to give 41 mg (11%) of 2-((1,1,2,2,2-pentamethyldisilyl)ethynyl)-9H-xanthen-9-one as a pale yellow powder, MP = 65–67 °C. 1H NMR (CDCl3; 300 MHz) δ 8.41 (d, J = 2.1 Hz, 1H); 8.32 (dd, J = 78.1 and 1.7 Hz, 1H); 7.78–7.71 (2H); 7.50–7.37 (3H); 0.30 (s, 6H), 0.19 (s, 9H). 13C NMR (CDCl3; 75 MHz) δ 176.4; 155.9; 155.5; 137.8; 135.0; 130.3; 126.8; 124.1; 121.6; 121.5; 119.5; 118.1; 118.0; 105.5; 94.3; −2.5; −3.1 (ESI†).
2-Isopropylthioxanthone (ITX from Lamberti Spa) is used as a reference photoinitiator. Diphenyliodonium hexafluorophosphate (Φ2I+), methyldiethanolamine (MDEA) and naphthalene were obtained from Aldrich. Trimethylolpropane triacrylate TMPTA and a bulk oligomer/monomer formulation based on 75/25 w/w epoxyacrylate/tripropyleneglycoldiacrylate (Ebecryl 605) were obtained from Cytec.
Photopolymerization
In film FRP experiments (60 µm thick films) under air, a given photoinitiator was dissolved in the polymerizable medium (Ebecryl 605 from Cytec). For TMPTA, 20 µm thick films were prepared. The irradiation conditions were similar to those described in detail in ref. 7: exposure to the non-filtered polychromatic light delivered by a Xe–Hg lamp (Hamamatsu, L8252, 150 W). The evolution of the double bond content was continuously followed by real-time FTIR spectroscopy at about 1640 cm−1 (Nexus 870, Nicolet) as described in ref. 7.For cationic polymerization experiments under air (∼20 µm thick films), weight concentrations of 1% in diphenyliodonium hexafluorophosphate (Φ2I+ from Aldrich) and 1% in PI were used to polymerize a di-(cycloaliphatic epoxide) monomer (Uvacure 1500 from Cytec) under a filtered light at λ > 400 nm or λ > 300 nm (see figure captions) delivered by a Xe lamp, Hamamatsu, L8253, 150 W. The evolution of the epoxy group content is continuously followed by real-time FTIR spectroscopy; the absorbance of the epoxy group was monitored at about 800 cm−1 as in ref. 8.
Computational procedure
Molecular orbital calculations were carried out with the Gaussian 03 suite of programs;9 the bond dissociation energy (BDE) (Si–Si) and the triplet energy level were calculated at the UB3LYP/6-31 + G* level. The different optimized geometries were frequency checked. The electronic absorption spectra for TX–Si and ITX were calculated with the time-dependent density functional theory at MPW1PW91/6-311++G** level on the relaxed geometries calculated at UB3LYP/6-31 + G* level.Laser flash photolysis
Nanosecond laser flash photolysis (LFP) experiments were carried out using a Q-switched nanosecond Nd/YAG laser (λexc = 355 nm, 9 ns pulses; energy reduced down to 10 mJ) from Continuum (Powerlite 9010) and an analyzing system consisting of a pulsed xenon lamp, a monochromator, a fast photomultiplier and a transient digitizer.10ESR spin trapping experiments
ESR spin trapping experiments (a well established technique for the identification of the radical center)11 were carried out using a X-Band spectrometer (MS 200 Magnettech). The radicals generated by light irradiation (Xe–Hg lamp—Hamamatsu, L8252, 150 W; λ > 310 nm) under argon were trapped by phenyl-N-tert-butylnitrone (PBN). The ESR spectra simulations were carried out with the PEST WINSIM program.12Results and discussion
(a) Photochemical properties of TX–Si and XT–Si
Interestingly (see Fig. 1A and B) the UV absorption of TX–Si is red-shifted compared to that of ITX (390 nm vs. 382 nm for TX–Si and ITX in acetonitrile, respectively; see also Fig. S1, ESI†). A similar red-shift is noted (Fig. 1C) for the derivative based on the xanthonechromophore (352 nm vs. 337 for XT–Si and XT in acetonitrile, respectively). A significant increase of the molar absorption coefficient ε (Fig. S1, ESI†) is also noted for TX–Sii.e. from 5200 M−1 cm−1 for ITX to 6000 M−1 cm−1; for XT–Si compared to XT, despite a red shifted absorption, a decrease of the absorption coefficient is found (from about 8000 M−1 cm−1 to about 5000 M−1 cm−1 for XT and XT–Si, respectively—Fig. 1C). The molecular orbitals involved in this near UV transition (HOMO → LUMO) are depicted in Fig. 2 for TX–Si. An interesting participation of the triple bond to the HOMO can be noted. For XT–Si, the same behavior is observed (Fig. S2 in the ESI†). Remarkably, for TX–Si, new intense absorption bands are also observed at 292 and 318 nm with ε = 42900 and 28500 M−1 cm−1, respectively (Fig. 1B). As deduced from MO calculations, these bands involve the HOMO − 1 and LUMO + 1 orbitals that are strongly delocalized on the alkynegroup (Fig. S2 in the ESI†).
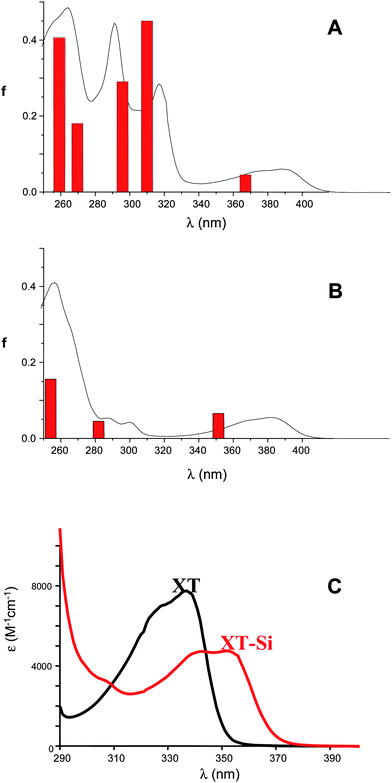 | ||
| Fig. 1 UV absorption properties. (A) TX–Si and (B) ITX in acetonitrile. The calculated absorption spectra (MPW1PW91/6-311++G** level—see text) are given (sticks). (C) UV absorption spectrum of XT–Si and XT in acetonitrile. | ||
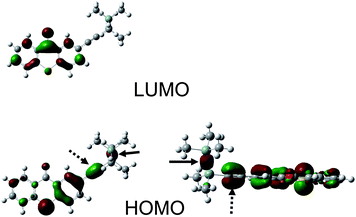 | ||
| Fig. 2 Highest occupied molecular orbital HOMO and lowest unoccupied molecular orbital LUMO for TX–Si. The triple bond is indicated by an arrow (dotted line); the Si–Si bond is indicated by another arrow (full line). | ||
In acetonitrile, the fluorescence maximum of TX–Si is located at 420 nm and the fluorescence lifetime was determined as 250 ps (using Fluoromax 4, Jobin-Yvon). A singlet state S1 energy level is calculated at 296 kJ mol−1 quite close to that of ITX (300 kJ mol−1). The fluorescence quantum yield was found similar to that of ITX. As in XT, for XT–Si, no fluorescence emission was detected. These results probably evidence the weak influence of the alkyne substituent on the S1 properties.
The photolysis of TX–Si is quite slow allowing an easy investigation of the excited state processes by laser flash photolysis. For XT–Si and for a similar light absorption than TX–Si, the very fast photolysis of the sample prevents a deeper investigation by LFP (Fig. 3). This also prevents the determination of the lifetime of 3XT–Sii.e. a strong photolysis is observed even with 5 laser shots. The excited triplet state T1 absorption of TX–Si is observed at 700 nm in acetonitrile (Fig. 4) compared to 650 nm for ITX. T1 is strongly quenched by O2 and naphthalene (well-known triplet energy acceptors); the interaction rate constants, gathered in Table 1, are >109 M−1s−1. The T1 lifetime is quite long (∼3 µs). The interaction rate constants of T1 with different additives (amine, onium salt) relevant for the polymerization processes are also reported in Table 1.
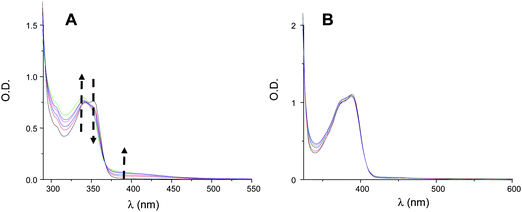 | ||
| Fig. 3 Photolysis of XT–Si (A) and TX–Si (B) for a similar light absorption in acetonitrile upon a laser irradiation at 355 nm (5 mJ per pulse); each spectrum is separated by 15 laser shots. | ||
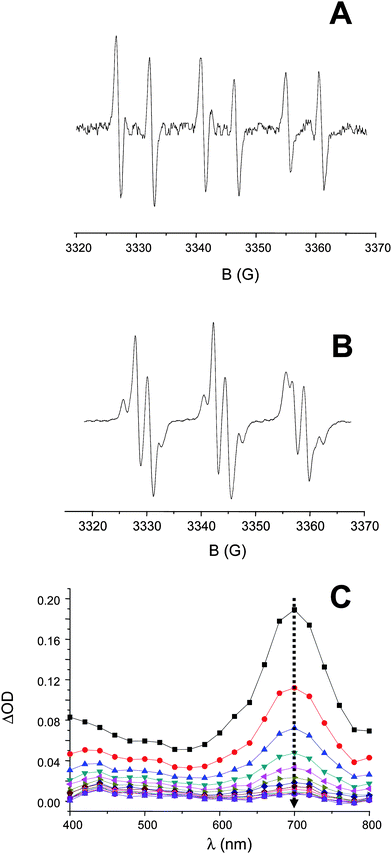 | ||
| Fig. 4 ESR spin trapping experiments of (A) TX–Si and (B) XT–Si (using PBN in tert-butylbenzene) under UV-light irradiation (λ > 300 nm) under argon; and (C) transient spectra of TX–Si in acetonitrile observed in LFP after laser excitation at 355 nm; from t = 0 to 23 µs by steps of 1.7 µs. | ||
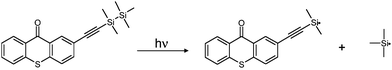
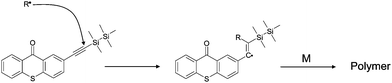
From ESR-ST experiments, the formation of silyl radicals upon irradiation is clearly evidenced for both TX–Si and XT–Si (Fig. 4) i.e. the hyperfine splitting (hfs) constants of the PBN adducts (aN = 14.5; aH = 5.6 G) being in full agreement with the presence of the known R3Si˙ radical.13 This supports a Si–Si bond cleavage of the disilyl moiety (Scheme 2). The calculated bond dissociation energy BDE (Si–Si) is quite low (evaluated here as ∼69 kcal mol−1 at UB3LYP/6-31 + G* level for both XT–Si and TX–Si). Such a cleavage process has been already observed in other ketones incorporating a Si–Si bond13b and in phenylethynyldisilanes.13c Interestingly (Fig. 4), in XT–Si, a carbon centered radical is also detected (aN = 14.5; aH = 2.3 G). This species can be ascribed to the vinyl radical generated by a silyl radical addition to the triple bond (Scheme 3): such a kind of addition is known.14
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
Using a similar LFP procedure as presented in ref. 15a, the addition rate constant of the tris(trimethylsilyl)silyl radical to cyclopentylacetylene in di-tert-butylperoxide was evaluated as 4.2 × 106 M−1s−1. This is in agreement with previous works which have evidenced the high reactivity of silyls towards alkynes.15b More detailed and highly useful investigations of silyl radicals reactivity are given in ref. 15. The low photolysis of TX–Si suggests a lower silyl radical formation ability in TX–Si compared to XT–Si: this also explains why vinyl radicals were not observed in the ESR-ST experiments on TX–Si.
A high interaction rate constant for the 1TX–Si/Φ2I+ interaction in acetonitrile was determined from fluorescence quenching (1.5 × 1010 M−1 s−1vs. 9.6 × 109 M−1s−1 for 1ITX/Φ2I+). The formation of Ph˙ was clearly supported by ESR-ST experiments (Fig. S3, ESI†) in full agreement with an electron transfer process (1). The corresponding amount of the R3Si˙ adduct is decreased by about 80% compared to similar experiments using TX–Si alone (see Fig. S3†).
| 1,3TX–Si+ Φ2I+ → TX–Si˙+ + ΦI + Φ˙ | (1) |
Taking into account the S1 lifetime and the iodonium salt quenching, one could expect a [R3Si˙] decrease of only ∼20% in TX–Si/Φ2I+ if the TX–Si cleavage occurs in the S1 state. On the other side, due to the long T1 lifetime and the efficient quenching of T1 by the iodonium salt (Table 1), a cleavage occurring from T1 alone would be more affected and [R3Si˙] strongly decreases down to ∼100%. Therefore, the experimental ESR-ST result (80% decrease) qualitatively suggests that the dissociation mainly occurs from T1, together with a minor contribution of a S1 cleavage.
Due to the similar fluorescence properties of TX–Si and ITX, the intersystem crossing quantum yields are expected to be closed. The cleavage rate constant in T1 is nevertheless rather low (kdiss < 3 × 105s−1) as the triplet state lifetime is relatively long. On the opposite, the shorter S1 lifetime of TX–Si (250 ps) compared to 2-methylthioxanthone (430 ps)2g could be in line with the minor concomitant S1 cleavage. For XT–Si, a triplet state cleavage should be likely involved as the intersystem crossing quantum yield for XT–Si is likely close to 1.0 (as for XT).16
The dissociation process is probably more efficient in XT–Si compared to TX–Si in agreement with the higher calculated triplet energy level of XT–Si (70 vs. 64 kcal mol−1) rendering the cleavage reaction energetically more favorable. With a BDE (Si–Si) ≈ 69 kcal mol−1, the dissociation is found slightly exothermic from 3XT–Si and endothermic from 3TX–Si. This result is fully consistent with both the faster photolysis of XT–Si mentioned above and the low T1 cleavage rate constant of 3TX–Si.
(b) Free radial photopolymerization FRP ability
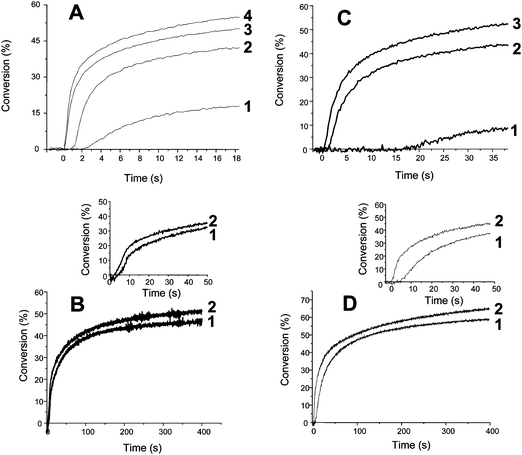 | ||
| Fig. 5 (A) Radical photopolymerization ability of (1) ITX (1% w/w), (2) TX–Si (1% w/w), (3) ITX/MDEA (1/1% w/w) and (4) TX–Si/MDEA (1/1% w/w) in an Ebecryl 605 film under air, Hg–Xe lamp exposure. (B) Cationic photopolymerization of Cyracure 6110 under air: in the presence of (1) ITX/Φ2I+ (1/1% w/w) and (2) TX–Si/Φ2I+ (1/1% w/w); xenon lamp exposure λ > 390 nm; inset: time range 0–50 s. (C) Radical photopolymerization ability of (1) XT (1% w/w); (2) XT–Si (1% w/w); and (3) XT–Si/MDEA (1/3% w/w) in an Ebecryl 605 film under air, Hg–Xe lamp exposure. (D) Cationic photopolymerization of Cyracure 6110 under air. In the presence of (1) XT/Φ2I+ (1/1% w/w) and (2) XT–Si/Φ2I+ (1/1% w/w). Xenon lamp exposure λ > 300 nm. Inset: time range 0–50 s. | ||
(c) Extractability of the photoinitiator
The extractability behavior has been checked (for demonstration) in the case of TX–Si using, first, similar conversions of Ebecryl 605 (about 70% after 400 s of irradiation with the Xe–Hg lamp) in the presence of the TX–Si/MDEA and ITX/MDEA photoinitiating systems. The extractability was evaluated from the amount of the photoinitiatior extracted with tetrahydrofuranTHF from the formed polymers. Interestingly, the extractability of TX–Si in our experimental conditions is 3 times lower than for ITX (Fig. 5, ESI†). Without MDEA, the extractability is quite similar for TX–Si/MDEA and TX–Si (only 10% higher for TX–Si) although the conversion is slightly lower for TX–Si (Fig. 5A). The Type I character of TX–Si is highly worthwhile i.e. in addition to the TX–Si trapped in the polymer network, TX fragments are also linked to the macromolecular chains (through reactions in Scheme 2). Moreover, from ref. 17 and our LFP result, the insertion of the alkyne fragment into the polymer network can contribute to some extent to the better behavior of TX–Si (Scheme 3 where R˙ can be an initiating or a polymerization propagating radical). This process is found favorable and exothermic for the addition of an acrylate radical to the alkynegroup of TX–Si by molecular orbital calculations (−37 kJ mol−1 at UB3LYP/6-31 + G* level).(d) Cationic photopolymerization (CP/FRPCP)
Fig. 5B and D show the relative efficiency of TX–Si/Φ2I+vs.ITX/Φ2I+ and XT–Si/Φ2I+vs.XT/Φ2I+ as cationic photoinitiating systems in aerated conditions. As aryl iodonium salts show a main absorption band at λ < 300 nm, the initiation ability of Φ2I+ alone in our irradiation conditions is negligible. XT–Si and TX–Si exhibit an almost similar polymerization initiating ability. Interestingly, TX–Si and XT–Si compared to ITX and XT lead to an increase of both the Rp and the final conversion (this increase is higher for the xanthone derivative). This can be explained on the basis of the two competitive initiation routes in TX–Si/Φ2I+ or XT–Si/Φ2I+: (i) a CP process where the photosensitized cleavage of the iodonium salt occurs according to eqn (1) and forms the TX–Si˙+ or XT–Si˙+ cation radicals (as ITX˙+ in ITX/Φ2I+) and (ii) a FRPCP process involving a direct cleavage of XT–Si and TX–Si generating silyl radicals (R3Si˙) easily oxidized by Φ2I+ and thereby leading to silylium cations eqn (2) that are known8,18 as highly efficient cationic initiating species. The oxidation of vinyl radicals (Scheme 3) to vinyl cations can also participate to some extend in the FRPCP process.19 The description of other FRPCP processes can be found for example in ref. 20 and references therein. The polymerization thus becomes a combination of CP with FRPCP. The balance between the two processes obviously qualitatively account for the relative efficiency increase of XT–Sivs.XT compared to TX–Sivs.ITX.| R3Si˙ + Φ2I+ → R3Si+ + ΦI + Φ˙ | (2) |
Conclusions
In the present paper, a novel cleavable thioxanthone photoinitiator TX–Si containing a disilylacetylene moiety leads to a quite interesting polymerization efficiency in FRP under air. This compound allows both to keep a recognized and largely used absorbing TXchromophore and to benefit from an unusual Type I behavior: this helps to decrease the PI migration outside the film, avoid the presence of any co-initiator and enhance the light absorption in the UV/visible wavelength range. In FRPCP under air, TX–Si is also a little better than ITX. XT–Si could also be an interesting compound exhibiting a better cleavage promising ability but still a lower UV/visible absorption. As demonstrated here, combining the thioxanthone/xanthone (or other PI skeletons) and silyl chemistries should open up a new promising direction of research.Acknowledgements
This work was supported by the “Agence Nationale de la Recherche” ANR under Grant ANR-10-BLAN-0802 (SILICIUM 2010).References
- (a) Photopolymerization: Fundamentals and Applications, ACS Symposium series, ed. A. B. Scranton, A. Bowman and R. W. Peiffer, Washington, DC, 1997, vol. 673 Search PubMed; (b) J. V. Crivello, Photoinitiators for Free Radical, Cationic and Anionic Photopolymerization, ed. G. Bradley, New York, 2nd edn, 1998 Search PubMed; (c) K. Dietliker, A Compilation of Photoinitiators Commercially Available for UV Today, Sita Technology Ltd, Edinburgh, London, 2002 Search PubMed; (d) Photoinitiated Polymerization, ed. K. D. Belfied and J. V. Crivello, ACS Symposium series, Washington, DC, 2003, vol. 847 Search PubMed; (e) Radiation Curing in Polymer Science and Technology, ed. J. P. Fouassier and J. F. Rabek, Elsevier Science Publishers Ltd, London, 1993 Search PubMed; (f) J. P. Fouassier, Photoinitiation, Photopolymerization and Photocuring: Fundamental and Applications, Hanser Publishers, New York, 1995 Search PubMed; (g) Photochemistry and UV Curing, ed. J. P. Fouassier, Research Signpost, Trivandrum, India, 2006 Search PubMed; (h) Basics of Photopolymerization Reactions, ed. J. P. Fouassier and X. Allonas, Research Signpost, Trivandrum, India, 2010, in press Search PubMed.
- (a) M. V. Encinas, A. M. Rufs, T. Corrales, F. Catalina, C. Peinado, K. Schmith, M. G. Neumann and N. S. Allen, Polymer, 2002, 43, 3909–3913 CrossRef CAS; (b) S. L. Herlihy, L. Shaun, PCT Int. Appl., WO 0372568, 2003, CA 139: 231973z; (c) M. Sabahi, E. L. Williams, W. Kuang, C. A. Brady, PCT Int. Appl., WO 2007 092935; (d) X. Jiang and J. Yin, Macromol. Chem. Phys., 2008, 209, 1593–1600 CrossRef CAS; (e) T. Corrales, F. Catalina, N. S. Allen and C. Peinado, in Photochemistry and UV Curing, ed. J. P. Fouassier, Research Signpost, Trivandrum, India, 2006 Search PubMed; (f) W. A. Green, in Basics of Photopolymerization Reactions, ed. J. P. Fouassier and X. Allonas, Research Signpost, Trivandrum, India, 2010, in press Search PubMed; (g) M. G. Neumann, M. H. Gehlen, M. V. Encinas, N. S. Allen, T. Corrales, C. Peinado and F. Catalina, J. Chem. Soc., Faraday Trans., 1997, 93, 1517–1521 RSC.
- (a) D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS; (b) G. Yilmaz, B. Aydogan, G. Temel, N. Arsu, N. Moszner and Y. Yagci, Macromolecules, 2010, 43, 4520–4526 CrossRef CAS; (c) B. Gacal, H. Akat, D. K. Balta, N. Arsu and Y. Yagci, Macromolecules, 2008, 41, 2401–2405 CrossRef CAS; (d) M. Aydin, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2005, 38, 4133–4138 CrossRef CAS; (e) L. Cokbaglan, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2003, 36, 2649–2653 CrossRef CAS; (f) H. Matsushima, S. Hait, Q. Li, H. Zhou, M. Shirai and C. E. Hoyle, Eur. Polym. J., 2010, 46, 1278–1287 CrossRef CAS.
- (a) T. Corrales, F. Catalina, C. Peinado, N. S. Allen, C. Bueno, A. M. Rufs and M. V. Encinas, Polymer, 2002, 43, 4591–4597 CrossRef CAS; (b) X. Jiang, H. Xu and J. Yin, Polymer, 2003, 45, 133–140; (c) X. Jiang and J. Yie, Macromol. Rapid Commun., 2004, 25, 748–752 CrossRef CAS; (d) T. Corales, F. Catalina, N. S. Allen and C. Peinado, J. Photochem. Photobiol., A, 2005, 169, 95–100 CrossRef; (e) G. Temel, N. Arsu and Y. Yagci, Polym. Bull., 2006, 57, 51–56 CrossRef CAS; (f) S. Hai, H. Zhou, H. Matsushima, C. E. Hoyle, Proceeding RadTech USA Chicago, 2008 Search PubMed; (g) X. Jiang, J. Luo and J. Yin, Polymer, 2009, 50, 37–41 CrossRef CAS; (h) Y. Wen, X. Jiang, R. Liu and J. Yin, Polymer, 2009, 50, 3917–3923 CrossRef CAS; (i) G. Temel, D. K. Balta, D. Sevinc and N. Arsu, in Basics of Photopolymerization Reactions, ed. J. P. Fouassier and X. Allonas, Research Signpost, Trivandrum, India, 2010, in press Search PubMed.
- (a) M. A. Tehfe, J. Lalevée, D. Gigmes and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1830–1837 CrossRef CAS; (b) M. El-Roz, J. Lalevée, X. Allonas and J. P. Fouassier, Macromolecules, 2009, 42, 8725–8732 CrossRef CAS; (c) M. A. Tehfe, J. Lalevée, D. Gigmes and J. P. Fouassier, Macromolecules, 2010, 43, 1364–1370 CrossRef CAS; (d) J. Lalevée, M. A. Tehfe, D. Gigmes and J. P. Fouassier, Macromolecules, 2010, 43, 6608–6615 CrossRef CAS.
- (a) H. Gilman and J. Diehl, J. Org. Chem., 1959, 24, 1914–1916 CrossRef CAS; (b) J. Smirnova, D. Wöll, W. Pfleiderer and U. E. Steiner, Helv. Chim. Acta, 2005, 88, 891–904 CrossRef CAS; (c) J. Zhao and R. C. Larock, J. Org. Chem., 2007, 72, 583–588 CrossRef CAS; (d) M. K. J. ter Wiel and B. L. Feringa, Tetrahedron, 2009, 65, 4332–4339 CrossRef CAS.
- (a) J. Lalevée, X. Allonas, S. Jradi and J. P. Fouassier, Macromolecules, 2006, 39, 1872–1879 CrossRef CAS; (b) J. Lalevée, L. Zadoina, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2494–2502 CrossRef CAS.
- M. El-Roz, J. Lalevée, F. Morlet-Savary, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7369–7375 CrossRef CAS.
- (a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 03, Revision B.2, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed; (b) J. B. Foresman and A. Frisch, Exploring Chemistry with Electronic Structure Methods, Gaussian. Inc., 2nd edn, 1996 Search PubMed.
- J. Lalevée, X. Allonas and J. P. Fouassier, J. Am. Chem. Soc., 2002, 124, 9613–9621 CrossRef CAS.
- (a) P. Tordo, Spin-trapping: Recent Developments and Applications, in Electron Spin Resonance, ed. N. M. Atherton, M. J. Davies and B. C. Gilbert, The Royal Society Of Chemistry, Cambridge, 1998, vol. 16 Search PubMed; (b) C. F. Chignell, Pure Appl. Chem., 1990, 62, 301–308 CrossRef CAS; (c) Y. Kotake and K. Kuwata, Bull. Chem. Soc. Jpn., 1981, 54, 394–399 CrossRef CAS; (d) A. Alberti, M. Benaglia, D. Macciantelli, S. Rossetti and M. Scoponi, Eur. Polym. J., 2008, 44, 3022–3027 CrossRef CAS.
- D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–112 CrossRef CAS.
- (a) H. Chandra, I. M. T. Davidson and M. C. R. Symons, J. Chem. Soc., Perkin Trans. 2, 1982, 1353–1356 RSC; (b) J. Lalevée, N. Blanchard, M. El-Roz, B. Graff, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 4180–4186 CrossRef CAS; (c) M. Ishikawa, H. Sugisawa, T. Fuchikami, M. Kumada, T. Yamabe, H. Kawakami, K. Fukui, Y. Ueki and H. Shizuka, J. Am. Chem. Soc., 1982, 104, 2872–2878 CrossRef CAS.
- D. L. Haire, U. M. Oehler, P. H. Krygsman and E. G. Janzen, J. Org. Chem., 1988, 53, 4535–4542 CrossRef.
- (a) J. Lalevée, X. Allonas and J. P. Fouassier, J. Org. Chem., 2007, 72, 6434–6439 CrossRef CAS; (b) C. Chatgilialoglu, in Organosilanes in Radical Chemistry, John Wiley & Sons, Chichester, 2004 Search PubMed; (c) J. Lalevée, A. Dirani, M. El-Roz, X. Alonas and J. P. Fouassier, Macromolecules, 2008, 41, 2003–2010 CrossRef CAS; (d) C. Chatgilialoglu, K. U. Ingold, J. Lusztyk, A. S. Nazran and J. C. Scaiano, Organometallics, 1983, 2, 1332–1335 CrossRef CAS; (e) C. Chatgilialoglu, J. C. Scaiano and K. U. Ingold, Organometallics, 1982, 1, 466–469 CrossRef CAS; (f) C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229–1251 CrossRef CAS; (g) C. Chatgilialoglu, V. I. Timokhin, A. B. Zaborovskiy, D. S. Lutsyk and R. E. Prystansky, Chem. Commun., 1999, 405–406 RSC.
- J. C. Scaiano, J. Am. Chem. Soc., 1980, 102, 7747–7753 CrossRef CAS.
- P. C. Montevecchi, M. L. Navachhia and P. Spagnolo, Tetrahedron, 1997, 53, 7929–7936 CrossRef CAS.
- (a) R. Souane, M. A. Tehfe, J. Lalevée, D. Gigmes and J. P. Fouassier, Macromol. Chem. Phys., 2010, 211, 1441–1445 CrossRef CAS; (b) J. Lalevée, X. Allonas and J. P. Fouassier, Chem. Phys. Lett., 2009, 469, 298–303 CrossRef CAS.
- Y. Yagci, C.-H. Fischer and W. Schnabel, Makromol. Chem. Rapid Commun., 1989, 10, 137–144 CrossRef CAS.
- (a) A. Ledwith, Polymer, 1978, 19, 1217–1219 CrossRef CAS; (b) H. Baumann and H. J. Timpe, Z. Chem., 1984, 24, 18–19 CAS; (c) C. Dursun, M. Degirmenci, Y. Yagci, S. Jockusch and N. J. Turro, Polymer, 2003, 44, 7389–7396 CrossRef CAS; (d) Y. Bi and D. C. Neckers, Macromolecules, 1994, 27, 3683–3693 CrossRef CAS; (e) J. V. Crivello, J. Macromol. Sci., Part A: Pure Appl. Chem., 2009, 46, 474–483 Search PubMed; (f) J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 866–875 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (i) UV absorption properties of TX–Si and ITX in tert-butylbenzene/acetonitrile (95%/5%); (ii) (a) HOMO − 1 and LUMO + 1 for TX–Si and (b) HOMO and LUMO for XT–Si; (iii) spin trapping experiments under the UV-light irradiation (λ > 300 nm) of TX–Si/Φ2I+ using PBN; (iv) radical photopolymerization ability of ITXvs.TX–Si in TMPTA; (v) photoinitiator extracted from the polymer with THF (storage of the polymer 48 h in this solvent); and (vi) NMR spectra for the investigated compounds. See DOI: 10.1039/c0py00392a |
| This journal is © The Royal Society of Chemistry 2011 |