How does a star chain (nanooctopus) crawl through a nanopore?
Hui Gea,
Stergios Pispasb and
Chi Wu†
*ac
aDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong
bTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vass. Constantinou Avenue, 11635, Athens, Greece
cThe Hefei National laboratory of Physical Science at Microscale, Department of Chemical Physics, The University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 14th January 2011
Abstract
The ultrafiltration of star-like polystyrene chains with different arm lengths (LA) and arm numbers (f) passing through a nanopore (20 nm) under an elongational flow field was investigated in terms of the flow-rate dependent relative retention ((C0 − C)/C0), where C0 and C are the polymer concentrations before and after the ultrafiltration. Our results reveal that for a given LA, the critical flow rate (qc,star), below which star chains are blocked, dramatically increases with f; but for a given f, is nearly independent on LA, contradictory to the previous prediction made by de Gennes and Brochard-Wyart. We have revised their theory in the region fin < fout, where fin and fout are the numbers of arms inside and outside the pore, respectively; and also accounted for the effective length of each blob. In the revision, we show that qc,star is indeed independent of LA but related to both f and fin in two different ways, depending on whether fin ≤ f/2 or ≥ f/2. A comparison of our experimental and calculated results reveals that most star chains pass through the nanopores with fin ∼ f/2. Further study of the temperature dependent (C0 − C)/C0 of polystyrene in cyclohexane shows that there exists a minimum of qc,star at ∼38 °C, close to the theta temperature of polystyrene star chains.
Introduction
De Gennes and Pincus predicted that the critical (minimum) flow rate (qc,1) for a linear chain to pass through a nanopore is independent of both the chain length and the pore size,1,2i.e., qc,l = kBT/(3πη), where kB, T and η are the Boltzmann constant, the absolute temperature and viscosity, respectively. Our previous study showed that the chain length indeed has no effect on qc,1,3 but qc,1 decreases as the pore size increases. In addition, the measured qc,1 is ∼10–200 time smaller than the predicted ones, depending on the solvent quality and the pore size. Such discrepancies are attributed to an over-simplified assumption in theory; namely, each blob (the subchain confined inside the nanopore) is a hard sphere so that its experienced hydrodynamic force along the flow direction is linearly proportional to the pore diameter (D). In reality, the hydrodynamic force (Fh) experienced by the subchain should be related to its effective length (Le) along the flow direction, i.e., the integration of all segments inside each subchain along the flow direction,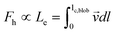 , where
, where 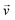 is the flow velocity, parallel to the central line of the nanopore and lc,blob is the contour length of the subchain. Assuming that the subchain inside each blob is a random Gaussian coil, the average projection of each segment with a length of ls along the flow direction is
is the flow velocity, parallel to the central line of the nanopore and lc,blob is the contour length of the subchain. Assuming that the subchain inside each blob is a random Gaussian coil, the average projection of each segment with a length of ls along the flow direction is 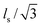 . Therefore,
. Therefore, 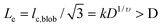 , where k is a scaling constant and 1/2 ≤ υ ≤ 3/5, depending on the solvent quality.4 Clearly, Fh is not a linear function of D so that qc,1 should be modified as:3
, where k is a scaling constant and 1/2 ≤ υ ≤ 3/5, depending on the solvent quality.4 Clearly, Fh is not a linear function of D so that qc,1 should be modified as:3
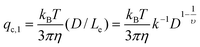 | (1) |
It is worth noting that on the basis of eqn (1), it is impossible to use the ultrafiltration of linear polymer chains through a nanopore to separate polymer chains with different lengths. The dependence of qc,1 on the pore size, was experimentally verified.3
In contrast to linear chains, the ultrafiltration of polymers with some complicated structure, such as star and branched configurations, through a nanopore is more intricate. In theory, a regular star polymer with f number of uniform arms joined at a central point might be the simplest case.5 In 1996, de Gennes and Brochard-Wyart6,7 formulated how such a star chain passes through a nanopore under an elongational flow field. They showed that the critical (minimum) flow rate (qc,star) depends not only on the total number of arms, but also on the number of forward arms (fin) squeezed into the nanopore. In their theory, qc,star is related to the arm length when fin < f/2, where they assumed that each forward arm inside the nanopore is fully stretched under the flow field, i.e., by the hydrodynamic force.
Such predictions and their speculated applications have existed for years,6–8 but have never been experimentally verified. This is partially because this kind of experimental study involves a combination of delicate polymer synthesis and precise physical characterization. Namely, 1) star polymers should be narrowly distributed in terms of both the arm number and length; 2) the hydrodynamic radius of stars must be larger than the pore diameter, i.e., each arm must be sufficiently long (over 103 monomers); and 3) the preparation of a proper nanopore structure so that the flow at its entrance is close to elongational without any rotational component.7 Recently, using high-vacuum anionic polymerization,9 we have overcome difficulties in the preparation of star chains with different arm numbers and lengths. Armed with these well-defined star polymers and using our previously established ultrafiltration method, we have finally experimentally studied how star chains pass through a nanopore.
Experimental section
Sample preparations
Polystyrene (PS) linear and star chains (up to 41 arms) with different lengths, as summarized in Table 1, were synthesized using a combination of high-vacuum anionic polymerization and a coupling method using divinylbenzene (DVB).9–11 Namely, using high-vacuum anionic polymerization, we first prepared narrowly distributed linear PS living chains with an active anionic end by initiating styrene (St, from Aldrich) with n-butyllithium (n-BuLi, 1.5M in cyclohexane, from Aldrich) in cyclohexane at ∼30 °C. Furthermore, these linear chains are coupled together to form star chains when a required amount of DVB molecules were added into the solution of living polystyl anions. The ratio of [DVB]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[anions] was kept at 0.7:1. All the chemicals were purified before the polymerization. The details can be found elsewhere.9,11 The coupling reaction between linear polystyl anions and DVB was lasted for 1–7 days, depending on the arm length. The longer the arm length was, the longer the coupling reaction time would be. Further addition of the same amount of DVB resulted in star chains with more arms. Such a step-by-step addition of DVB finally led to a series of star chains with different arm numbers but an identical arm length, as shown in Fig. 1.10
:[anions] was kept at 0.7:1. All the chemicals were purified before the polymerization. The details can be found elsewhere.9,11 The coupling reaction between linear polystyl anions and DVB was lasted for 1–7 days, depending on the arm length. The longer the arm length was, the longer the coupling reaction time would be. Further addition of the same amount of DVB resulted in star chains with more arms. Such a step-by-step addition of DVB finally led to a series of star chains with different arm numbers but an identical arm length, as shown in Fig. 1.10
| Code | f | M w,arm /(g mol−1) | M w,star/(g mol−1) | M w/Mn | <Rh>/nm |
|---|---|---|---|---|---|
| a Note that relative errors of Mw and <Rh> are 5% and 2%, respectively. | |||||
| Star-41 | 41 | 2.10 × 105 | 8.61 × 106 | 1.20 | 61 |
| Star-6A | 6 | 1.30 × 105 | 7.80 × 105 | 1.18 | 28 |
| Star-6B | 6 | 2.10 × 105 | 1.26 × 106 | 1.20 | 41 |
| Star-6C | 6 | 3.50 × 105 | 2.10 × 106 | 1.20 | 56 |
| Star-3 | 3 | 2.10 × 105 | 6.30 × 105 | 1.08 | 32 |
| Star-2 linear chain | 2 | 2.95 × 105 | 5.90 × 105 | 1.01 | 22 |
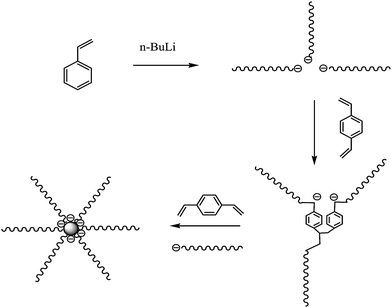 | ||
| Fig. 1 Schematic of the synthesis of star chains with different arm numbers but an identical arm length using anionic polymerization and the DVB coupling method. | ||
Note that such prepared star chains are normally broadly distributed in the arm number. Therefore, we have to further fractionate them by the following procedure;12 1) dissolving these star chains in 1,4-dioxane at 80 °C with a concentration of 0.03 g mL−1; 2) adding ethanol dropwise until the solution just became slightly milky; 3) letting the solution stand at the room temperature for few weeks so that a small fraction of star chains with the highest number of arms precipitated out of the solution; and 4) repeating steps 2 and 3 to obtain narrowly distributed star chains with different arms. The 3-arm star chains (Mw_arm = 2.1 × 105 g mol−1, Mw/Mn = 1.08) was previously synthesized by using chlorosilane as the coupling agent.9 The weight-average molar mass (Mw) and polydispersity index (Mw/Mn) of such obtained star chains were characterized by laser light scattering, also as summarized in Table 1.
Laser light scattering (LLS)
A modified commercial LLSspectrometer (ALV/DLS/SLS- 5022F) equipped with a multi-τ digital time correlator (ALV5000) and a cylindrical 22 mW UNIPHASE He–Ne laser (λ0 = 632 nm) was used. The incident beam was vertically polarized with respect to the scattering plane. The details of the LLS instrumentation and theory can be found elsewhere.13 Briefly, in static LLS, the excess absolute time-averaged scattered light intensity, known as the excess Rayleigh ratio Rvv(θ), of a dilute polymer solution at concentration C (g mL−1) is related to the weight average molar mass Mw, the root-mean square radius of gyration <Rg2>1/2, and the scattering vector ks as
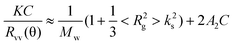 | (2) |
Ultrafiltration
In our ultrafiltration experiments, a double layer membrane filter (Whatman, Anotop 10) was used. The top and bottom layers contain an equal number of 200 nm and 20 nm cylindrical pores, respectively; i.e., each large pore is on top of a small pore. Such a structure prevents any possible interference of the flow fields generated by different small pores at their entrances. In each solution, we added a certain amount of short linear chains with a size smaller than the pore diameter. These short linear chains will pass through the nanopore even without any flow so they served as an internal standard. The concentrations of large star and short linear chains (CL and CS) are properly chosen so that <IL>/<IS> = CLML/CSMS ∼ 1, where ML and MS are the molar masses of large star and short linear chains, respectively. Note that in dynamic LLS, <IL>/<IS> equals the area ratio of their corresponding peaks in G(Γ). Since the nanopore has no retention on short linear chains, the decrease of <IL>/<IS> must be related to the retention of large star chains. On the other hand, in static LLS, we can measure the total time-average scattered light intensity (<Itot> = <IL> + <IS>). A combination of static and dynamic LLS results enables us to determine <IL> and <IS> and then the relative retention [(C0 − C)/C0] of larger star chains under different flow rates (q).15 In each ultrafiltration experiment, the solution temperature and q were controlled by an incubator (Stuart Scientific, S160D) (± 0.1 °C) and a syringe pump (Harvard Apparatus, PHD 2000), respectively.Result and discussion
Fig. 2 shows the flow rate (q) dependent relative retention of 6-arm star chains with different arm lengths in toluene. There is no obvious difference in “[(C0 − C)/C0] vs. q” among these different 6-arm star chains. As discussed in the introduction, no arm-length dependence of qc,star should only occur in the symmetrical mode;6 namely, fin = f/2. It has to be stated that de Gennes and Brochard-Wyart6 assumed that when 1 ≤ fin < f/2, each arm inside the nanopore is fully stretched so that its effective length along the flow direction is related to the contour length of each arm. In this way, star chains with shorter arms require a higher flow rate to pull the backward fout arms through the nanopore because the hydrodynamic force on each arm Fh is proportional to both the arm length and the flow rate. Apparently, if de Gennes and Brochard-Wyart were right, Fig. 2 would indicate that these 6-arm star chains would only pass through the nanopore via the symmetrical mode, i.e., fin = f/2.![Flow rate (q) dependence of relative retention [(C0 − C)/C0] of star chains with six arms but different arm lengths in toluene, where C0 and C are the polymer concentrations before and after the ultrafiltration.](/image/article/2011/PY/c0py00361a/c0py00361a-f2.gif) | ||
| Fig. 2 Flow rate (q) dependence of relative retention [(C0 − C)/C0] of star chains with six arms but different arm lengths in toluene, where C0 and C are the polymer concentrations before and after the ultrafiltration. | ||
Fig. 3 shows that the relative retention of star chains decreases as the flow rate increases. The change is apparently not as abrupt as the first-order coil-to-stretch transition of linear polymer chains observed before.3,15 It should be noted that here the x-axis is in a logarithmic scale. The transition is actually sharp except for star chains with 41 arms. For Star-41, fin might vary in the range 1–41 and each fin leads to one different qc,star, explaining why the change of “ relative retentionvs. q” is not as sharp as those for Star-6 and Star-3 with the same arm length.
![Flow rate (q) dependence of relative retention [(C0 − C)/C0] of star chains with different arm numbers but an identical arm length in toluene, where the weight-average molar mass (Mw,arm) of each arm is 2.1 × 105 g mol−1.](/image/article/2011/PY/c0py00361a/c0py00361a-f3.gif) | ||
| Fig. 3 Flow rate (q) dependence of relative retention [(C0 − C)/C0] of star chains with different arm numbers but an identical arm length in toluene, where the weight-average molar mass (Mw,arm) of each arm is 2.1 × 105 g mol−1. | ||
Fig. 4 shows a better view of a typical variation of the relative retention with q for Star-41, where we define two critical flow rates (qc,star,s and qc,star,p) marked the starting and peaking positions of the relative retention. For Star-41, qc,star,s = 2.40 × 10−12 and qc,star,p = 5.14 × 10−13 mL s−1. These measured critical flow rates are very different from those predicted by de Gennes and Brochard-Wyart.6 A combination of our experimental results in Fig. 2 and 3 forces us to rethink whether there is something missing/improper in their assumptions and theoretical treatments presented for a long time but never having been seriously tested.
![Differentiation of relative retention to flow rate, d[(C0 − C)/C0]/dq, for star chains with 41 arms, where qc,star,s and qc,star,p define the starting and peaking points of retention, respectively.](/image/article/2011/PY/c0py00361a/c0py00361a-f4.gif) | ||
| Fig. 4 Differentiation of relative retention to flow rate, d[(C0 − C)/C0]/dq, for star chains with 41 arms, where qc,star,s and qc,star,p define the starting and peaking points of retention, respectively. | ||
Fig. 5 schematically illustrates two different paths: 1 ≤ fin ≤ f/2 and f/2 ≤ fin ≤ f, depending on whether more arms are forward or backward when each star chain enters the nanopore. First, let us consider the translocation without any flow. If fin arms are forward and fill up the nanopore and each arm inside is made of nb blobs and each blob has a diameter of ξin, we have ξin2fin = D2 or ξin = D/fin1/2, as shown in Fig. 5. Thermodynamically, the energy and force to confine each blob is kBT and kBT/ξin, respectively, so that the total confinement energy and force (Ec and Fc) should be kBTnbfin and (kBT/ξin)nbfin. Assuming that each arm (blob) contains Narm (Nb) segments, nb = Narm/Nb and ξin = kNbν. Putting everything together, we have Ec = kBTNarm(k/D)1/νfin(1+1/2ν) for fin forward arms. If the entire star chain enters the nanopore, the total confinement energy should also include those backward (f − fin) arms, i.e., Etotal = kBTNarm(k/D)1/ν[fin(1+1/2ν) + (f − fin) (1+1/2ν)]. The differentiation dEtotal/dfin = 0 leads to that the minimum of Etotal occurs at fin = f/2.
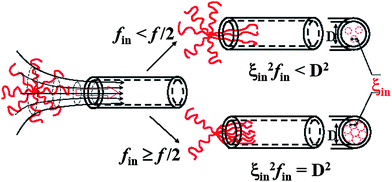 | ||
| Fig. 5 Schematic of how a star chain enters a nanopore under two different situations; namely, f ≥ fin ≥ f/2 and f/2 ≥ fin ≥ 1. | ||
Second, let us consider the translocation under a flow. When f/2 ≤ fin ≤ f, we only need to consider under which flow rate fin arms can enter the nanopore with a diameter (D), and do not need to worry about the backward arms because fout = f − fin < fin. The hydrodynamic force on each blob inside is 3πηle(q/D2) and the total hydrodynamic force (Fh) is 3πηle(q/D2)nbfin, where le is the effective length of each blob along the flow direction. The condition of Fh = Fc leads to the critical flow rate for f/2 ≤ fin ≤ f,
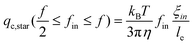 | (3) |
An attentive reader might find that in the above discussion we only need to consider under which flow rate the first blobs of fin forward arms can enter the nanopore because the second and remaining blobs of each arm just follow its first blob. When 1 ≤ fin ≤ f/2, we have to consider how many number of arms are outside of the nanopore, as shown in Fig. 5. Obviously, qc,star calculated in eqn (3) can only pull fin arms in, not sufficient to pull fout arms into the nanopore because fout > fin. Therefore, one has to increase q to stretch each forward arm inside the nanopore further, i.e., decreases ξin at the same time. In this way, ξin2fin becomes smaller than D2, as also shown in Fig. 5. The decrease of ξin leads to the increase of the force required to confine each blob. Finally, when 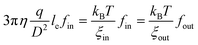 , the hydrodynamic force is just sufficient to pull the outside backward fout arms into and through the nanopore, wherein ξout2fout = D2, i.e., the nanopore is just filled with fout number of arms. Therefore, we have
, the hydrodynamic force is just sufficient to pull the outside backward fout arms into and through the nanopore, wherein ξout2fout = D2, i.e., the nanopore is just filled with fout number of arms. Therefore, we have
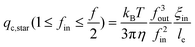 | (4) |
If each blob is considered as a hard sphere, i.e., le = ξin, eqn (3) and (4) become
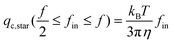 | (5a) |
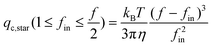 | (5b) |
Our revised formulas reveal that qc,star is independent of the arm length no matter whether fin is larger or smaller than f/2. Note that eqn (5a) is identical to that in ref. 6 in which de Gennes and Brochard-Wyart mistakenly wrote fin2 instead of fin.
The continuous lines in Fig. 6 shows how qc,star changes with fin for star chains with different arm numbers on the basis of eqn (5a) and (5b). Note that qc,star linearly increases with fin in the range f/2 ≤ fin ≤ f, but dramatically increases as fin decreases when fin ≪ f/2. The minimum is located at fin = f/2, different from the optimal fin predicted by de Gennes and Brochard-Wyart.6 Physically, this is reasonable because the decrease of fin reduces the overall hydrodynamic force on the forward arms inside the nanopore, and at the same time, increases the force required to confine those backward arms still outside the nanopore. As discussed in the section of Introduction, for linear chains, we can treat the subchain inside each blob as a random Gaussian coil so that le = kξin1/υ. Therefore, eqn (5a) and (5b) can be rewritten as
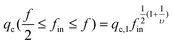 | (6a) |
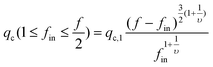 | (6b) |
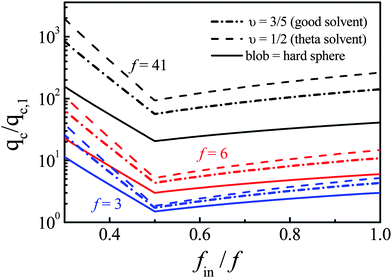 | ||
| Fig. 6 Forward arm number (fin) dependence of reduced critical flow rate (qc,star/qc,1) of star chains with different arm numbers but an identical arm length, calculated on the basis of eqn (5) and (6), respectively. | ||
For Star-41, qc,star,p/qc,1 is around 26, not too far away from the minimum point (fin = f/2) calculated from eqn (5), but lower than that calculated from eqn (6). The difference between eqn (5) and (6) is whether we should treat each blob as a hard sphere. When a half number of arms of a Star-41 chain is squeezed into a nanopore with a diameter of 20 nm, ξin is only 4–5 nm so that each arm should be highly stretched. Note that the estimated diameter of a polystyrene chain is ∼1.2 nm. Therefore, each blob can be viewed as a hard ball filled with the segments of the subchain without draining, explaining why its qc,star,p/qc,1 is close to those calculated from eqn (5). Also note that for Star-41, fin is larger than 13, which is understandable because qc,star increases sharply when fin is low for star chains with a high number of arms.
On the other hand, for Star-6 and Star-3, qc,star,p/qc,1 = 6.3 and 2.8 respectively, slightly higher than the minimum points (3 and 1–2) calculated based on eqn (5), but more close to 4 and 2 estimated from eqn (6). It indicates that for star chains with a lower number of arms, eqn (6) is a better choice because the subchain inside each blob is much less confined; namely, it is not necessary to fully stretch each arm to pull the outside backward arms into the nanopore. Also note that a comparison of experimentally measured values of qc,star/qc,l and eqn (6) shows that 2 ≤ fin(Star-6) ≤ 6 and 1 ≤ fin(Star-3) ≤ 3, revealing that star chains with a lower number of arms can enter the nanopore in different ways, even if its mostly preferred path is fin ∼ f/2.
Further, we studied the effect of the arm conformation on qc,star by dissolving polystyrene star chains in cyclohexane in which each arm contracts as the solution temperature decreases below its theta temperature (∼34.5 °C), as shown in Fig. 7. It should be noted that both the average radius of gyration (<Rg>) and hydrodynamic radius (<Rh>) of Star-41 chains decrease with the solution temperature, but the change is much smaller than that of linear PS chains within the same temperature range,5,16 presumably because 41 arms are crowed within a small space and the excluded volume prevents their collapse even in a poor solvent (lower temperatures). It should be noted that in the temperature range studied there is no change in the scattered light intensity, implying that there is no interchain association.
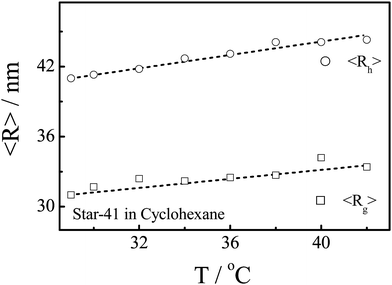 | ||
| Fig. 7 Solution temperature dependence of average radius of gyration (<Rg>) and average hydrodynamic radius (<Rh>) of star chains with 41 arms in cyclohexane. | ||
It has been previously suggested that the temperature at which qc reaches its minimum could be attributed to the true theta solvent,3 which was based of a reasonable assumption that polymer chains at the theta solvent are softest and mostly deformable, i.e., in a good solvent, the chain is highly swollen so that a stronger hydrodynamic force is required to stretch it into a string of blobs and confine each blob inside the nanopore due to its entropic elasticity; while in a poor solvent, the segment-segment attraction is stronger than the segment-solvent interaction so that a stronger hydrodynamic force is needed to overcome the enthalpy-originated force.
Fig. 8 shows the temperature dependence of qc,star,s and qc,star,p of Star-41 in cyclohexane at different flow rates. Despite that there is not much change in both <Rg> and <Rh>, qc,star varies a lot in the same temperature range because the collapse of a star chain at lower temperatures is hindered by the excluded volumes of different arms but the hydrodynamic force experienced inside the nanopore is related to the properties of individual arms, similar to that of linear chains free in solution. For Star-41, the minimum of qc is located at ∼38 °C, few degrees higher than that of linear polystyrene chains.17 Such a difference has been previously reported for star and branched chains, especially when the arm number is high and the arm or branch is long,18–20 presumably because the star and branching configurations increase the distance between segments so that the inter-segment attraction becomes stronger.
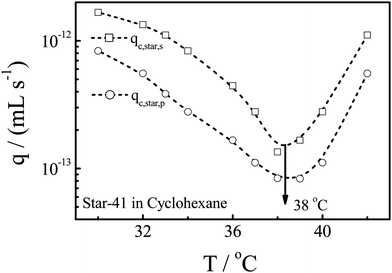 | ||
| Fig. 8 Solution temperature dependence of qc,star,s and qc,star,,p of star chains with 41 arms in cyclohexane. | ||
Conclusion
The ultrafiltration of star chains with different arm numbers and lengths reveals that the critical (minimum) flow rate (qc,star), at which the chains start to pass through a nanopore, is independent of the arm length but strongly influenced by the number of total arms and forward arms that initially enter the nanopore (f and fin), contrary to a previous prediction made by de Gennes and Brochard-Wyart in the 90s in which there exists an optimum number of fin between 1 and f/2. Our revision of their theory shows that such a discrepancy is attributed to their assumption that each forward arm inside the nanopore is fully stretched by the flow when fin < fout. In our revised formulation, the passing of a star chain through a nanopore depends on whether fin ≤ f/2 or fin ≥ f/2. In the case of fin ≥ f/2, qc,star linearly increases with fin, fairly slowly; but when fin ≤ f/2, qc,star ∼ (f − fin)3/fin2, it dramatically drops as fin increases, especially when f is high. The minimum of qc,star is exactly located at fin = f/2, independent of the arm number and length. Our experimental results confirm that most of star chains indeed pass through the nanopore with fin around f/2, especially when f is high, not involving the fully stretched forward arms. Further, our study of star chains with 41 arms in cyclohexane at different temperatures reveals that there is a minimum qc,star located at ∼38 °C, slightly higher than the theta temperature, which demonstrates that as in the case of linear chains, the ultrafiltration of star chains in a dilute solution through a nanopore provides a better and convenient way to determine the theta temperature. This study has laid a foundation for further applications of using the ultrafiltration to separate and characterize star chains with different arm numbers. Our results also have some implications in the design of non-viral polymeric carriers with different architectures for transporting drugs or genes into or through some organs, such as the kidney and liver.Acknowledgements
The financial support of the National Natural Scientific Foundation of China Projects (50773077 and 20934005) and the Hong Kong Special Administration Region Earmarked Projects (CUHK4037/07P, 2160331; CUHK4046/08P, 2160365; CUHK4039/08P, 2160361; and CUHK4042/09P, 2160396) is gratefully acknowledged.References
- P. G. De Gennes, J. Chem. Phys., 1974, 60, 5030 CrossRef CAS.
- P. Pincus, Macromolecules, 1976, 9, 386 CrossRef CAS.
- H. Ge, F. Jin, J. F. Li and C. Wu, Macromolecules, 2009, 42, 4400 CrossRef CAS.
- Polymer Solutions: An Introduction to Physical Properties, ed. I. Teraoka, John Wiley & Sons, Inc., New York, 2002 Search PubMed.
- M. Daoud and J. P. Cotton, J. Physique, 1982, 43, 531 Search PubMed.
- F. Brochard-Wyart and P. G. De Gennes, C. R. Acad. Sci., Ser. II, 1996, 323, 473 Search PubMed.
- P. G. De Gennes, Adv. Polym. Sci., 1999, 138, 91 CAS.
- D. M. Meunier and P. B. Smith, Macromolecules, 2005, 38, 5313 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747 CrossRef CAS.
- H. J. Lee, K. Lee and N. Choi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 870 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, S. Pispas and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3211 CrossRef CAS.
- S. Q. Zhou, S. Y. Fan, S. T. F. Au-yeung and C. Wu, Polymer, 1995, 36, 1341 CrossRef CAS.
- B. Chu. Laser Light Scattering, Academic Press,New York, 2nd edn, 1991 Search PubMed.
- B. J. Berne, R. Pecora. Dynamic Light Scattering, Plenum Press, New York, 1976 Search PubMed.
- F. Jin and C. Wu, Phys. Rev. Lett., 2006, 96, 237801 CrossRef.
- B. Appelt and G. Meyerhoff, Macromolecules, 1980, 13, 657 CrossRef CAS.
- B. Chu, Q. Ying and A. Y. Grosberg, Macromolecules, 1995, 28, 180 CrossRef CAS.
- D. Papanagopoulos and A. Dondos, Eur. Polym. J., 2004, 40, 2305 CrossRef CAS.
- F. Ganazzoli and G. Allegra, Macromolecules, 1990, 23, 262 CrossRef CAS.
- A. Dondos and D. Papanagopoulos, Polymer, 1997, 38, 6255 CrossRef CAS.
Footnote |
| † The Hong Kong address should be used for all correspondence. |
| This journal is © The Royal Society of Chemistry 2011 |