Selective cleavage of polymer grafts from solid surfaces: assessment of initiator content and polymer characteristics†
Susanne
Hansson
,
Per
Antoni
,
Helena
Bergenudd
and
Eva
Malmström
*
KTH Royal Institute of Technology, School of Chemical Engineering, Dept. of Fibre and Polymer Technology,
SE-100 44, Stockholm, Sweden. E-mail: mavem@kth.se; Fax: +468 790 8283; Tel: +468 790 7225
First published on 17th December 2010
Abstract
A novel initiator for atom transfer radical polymerization, also allowing for selective cleavage of polymer grafts, was designed and immobilized on a solid substrate. After cleavage, the initiator content was determined by utilizing Ellman's reagent and the cleaved polymer grafts were isolated and characterized by size exclusion chromatography.
With the aim to design new materials that meet our requirements for environmental awareness, the interest in cellulose has increased enormously during the last decades. To expand the applicability of cellulose-based materials, e.g. composites or more technologically advanced applications such as sensors, it is often essential to modify the cellulose to control or tailor the properties. This can be accomplished by grafting various polymers from the cellulose surface utilizing controlled polymerization methods, tailoring graft length by aiming for different ratios of monomer to sacrificial initiator. The sacrificial initiator forms unbound polymer that easily can be analyzed with respect to the molecular weight and polydispersity, as opposed to the grafted polymers that are covalently attached to the surface.
Atom transfer radical polymerization (ATRP) is a popular method employed when grafting polymers from various substrates, since it is a versatile and robust technique that provides excellent control over the grafting system.1–9 The disadvantages, however, are the relatively large amount of the transition metal catalyst required for control and the high sensitivity towards oxygen. To overcome or circumvent these drawbacks, activators regenerated by electron transfer (ARGET) ATRP can be employed.10 Our group has previously shown that this technique can be straightforwardly employed to graft a range of monomers from cellulose.11
However, to further understand the graft properties, detachment of the covalently linked polymer chains and their ensuing analyses are vital, along with assessment of initiator content. Zoppe et al.12 cleaved poly(N-isopropylacrylamide) grafts from cellulose nanocrystalsvia saponification (2% aq NaOH solution, 48 h), breaking the ester linkages of the immobilized initiators. Consequently, this method becomes limited to grafting polymers without sensitive groups, such as esters. Acidic hydrolysis of cellulose has also been attempted as a method to isolate the grafts for subsequent characterization, resulting in a complete degradation of the substrate.13–16 Zhou et al.17 cleaved poly(methyl methacrylate) (PMMA) by acidic hydrolysis (1 mM HCl(aq) overnight) from the surface of a xyloglucan layer that was adsorbed onto a filter paper prior to grafting. They concluded that the polymer growing from the surface has similar characteristics to the free polymer propagating from the sacrificial initiator occurring parallel to grafting, a statement also supported by others.9,18 Nonetheless, no information regarding the number of polymer chains on the surface was reported.
The purpose of the present research is to design an initiator that allows cleavage of the grafts as well as an estimation of the initiator content. In order to avoid degradation of sensitive substrates, like cellulose, mild reaction conditions are required, especially to allow subsequent surface analysis.
A disulfide containing initiator, inspired by the work of Matyjaszewski et al.,19–22 was designed and synthesized by reacting 2,2′-dithiodiethanol with α-bromoisobutyryl bromide. This product was reacted with succinic anhydride to introduce a carboxylic acid moiety and subsequently transformed into an acid chloride using oxalyl chloride. The acid chloride was reacted with the available hydroxyl groups on a cellulose substrate, Whatman No. 1 filter paper (2 × 3 cm2) was used for the immobilization. Morandi et al.5 showed that the initiator content on cellulose nanocrystals could be varied by changing the reaction time, the amount of the initiator, and the reaction temperature. Thus, the reaction time was varied: 0.25, 0.50, 1.0, and 15 h at room temperature.
The initiator-functionalized substrate was immersed into a mixture containing MMA, anisole, sacrificial initiator1 (Scheme S1†), N,N,N′,N′,N′-pentamethyldiethylenetriamine (PMDETA), Cu(II)Br2, and ascorbic acid, targeting a final degree of polymerization (DP) of 800, at full conversion. The free polymer formed from the sacrificial initiator was precipitated and thereafter analyzed with SEC, see Table 1.
The grafted cellulose substrate was thoroughly washed to remove any unbound polymeric residues. The successful grafting was confirmed by FT-IR analysis with the appearance of the characteristic peak at 1730 cm−1, originating from the carbonyl group (Fig. S1, ESI†).
The PMMA-grafted cellulose surface was subjected to a solution of 1,4-dithiothreitol (DTT) and triethylamine (TEA) in THF, in order to reduce the disulfide linker, detaching the grafted polymer (Scheme 1). FT-IR analyses of PMMA-grafted substrates demonstrated that most polymer chains were cleaved after only 18 h, suggesting that the disulfide bonds are readily accessible. However, to obtain complete cleavage, which is crucial for the following analysis, longer reaction times combined with a larger amount of DTT were required (Fig. S2–S4†).
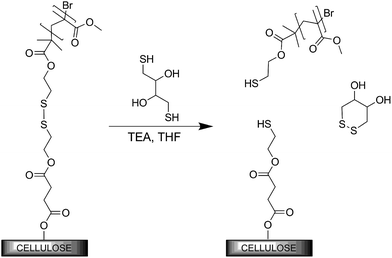 | ||
| Scheme 1 Selective graft cleavage of the PMMA-modified cellulose substrate, leaving the substrate intact. | ||
The substrate was carefully rinsed to collect all cleaved polymer. The amount of obtained polymer was insufficient for precipitation, giving rise to the SEC analyses of crude samples, see Table 1. The molar masses of the free and the cleaved polymers are in good agreement, suggesting that the polymerization occurs at a similar rate from the surface as from the sacrificial initiator, corroborating the hypothesis that grafts can be tailored by employing a sacrificial initiator. The slightly higher molar masses of the free polymers are probably due to slight fractionation during precipitation, resulting in an overall slightly higher average molar mass and lower PDI.
Field emission scanning electron microscopy (FE-SEM) was employed to study the surface topology. The fine structure of the initiator-functionalized cellulose substrate is apparent in Fig. 1a. Conversely, the PMMA-grafted cellulose substrate in Fig. 1b displays a smoother surface. After the PMMA-grafts have been cleaved from the surface, the fine structure of cellulose can be perceived again (Fig. 1c), indicating that the grafts have been successfully removed. The initiator-functionalized and DTT-treated cellulose structures are very similar to the unmodified substrate, establishing that both treatments are non-destructive.
 | ||
Fig. 1
FE-SEM micrographs of cellulose substrate (a) initiator-functionalized, (b) PMMA-grafted, and (c) after cleavage, magnified 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. 000×. | ||
The reduction of disulfide bonds uncovers thiol moieties on the surface readily quantifiable by Ellman's reagent23,24 (5,5′-dithiobis-2-nitrobenzoic acid (DTNB)). The reaction between DTNB and a thiol generates the chromophore 5-thio-2-nitrobenzoic acid (TNB), which absorbs strongly at 412 nm. The combination of the complete reduction of disulfide linkers and the quantitative nature of Ellman's reagent provides the possibility to estimate the initiator content of the substrate.
The DTT-treated substrate was immersed in a DTNB phosphate buffer solution, pH 7.0. An aliquot of the solution was characterized by UV analysis. The measured absorbance was used to calculate the number of moles of thiols present on the substrate (Table 2 and Fig. S5†). The graft density was estimated by employing the macroscopic surface area, considering the substrate as 2-D. However, due to the inherent surface roughness of cellulose, see Fig. 1, the true surface area, where porosities and irregularities are taken into account, was difficult to measure accurately. Consequently, the values for graft densities in Table 2 can only be considered as very rough estimates. Therefore, the initiator content was determined by normalizing the amount of thiols to the sample weight (Table 2), obtaining a better assessment that also is more applicable to use for different types of solid surfaces. As a comparison, the initiator content of silica nanoparticles was reported to be in the region of 70 to 250 µmol per g,9 suggesting that the values in Table 2 are reasonable for substrates with a low surface-area, such as the filter paper. To further verify the versatility of this method, microcrystalline cellulose (MCC)—with a diameter of 20 µm—was employed as an additional substrate, Table 2. The initiator content was found to be roughly twice as high as for filter papers.
| Samplea | Immob./h | Abs. (λ = 412 nm) | n thiols/µmol | Sample weight/mg | Graft densityb/nmol cm−2 | Initiator content/µmol g−1 |
|---|---|---|---|---|---|---|
| a Initiatory where y represents the immobilization time of the initiator. b Calculation based on the macroscopic area: 12 cm2. | ||||||
| Initiator0.25 cleaved | 0.25 | 0.653 ± 0.003 | 1.005 ± 0.004 | 53.9 | 83 | 18.6 ± 0.1 |
| Initiator0.5 cleaved | 0.50 | 0.686 ± 0.002 | 1.055 ± 0.003 | 51.2 | 88 | 20.6 ± 0.1 |
| Initiator1.0 cleaved | 1.0 | 0.681 ± 0.003 | 1.048 ± 0.005 | 52.0 | 88 | 20.2 ± 0.1 |
| PMMA240 − cleaved | 15 | 0.685 ± 0.001 | 1.053 ± 0.001 | 51.1 | 88 | 20.6 ± 0.1 |
| PMMA432 − cleaved | 15 | 0.701 ± 0.005 | 1.078 ± 0.008 | 51.3 | 90 | 21.0 ± 0.2 |
| MCC − cleaved | 3.5 | 0.661 ± 0.003 | 1.017 ± 0.004 | 25.6 | — | 39.7 ± 0.2 |
The results in Table 2 also show that only a short reaction time is required to convert the available hydroxyl groups on the surface into initiating moieties at room temperature. After 30 min of immobilization, the initiator content on the surface is essentially the same as after 15 h. A large surplus of the immobilized initiator was used, 1.33 mmol, compared to the number of reacted hydroxyl groups on the surface, ∼1 µmol, suggesting that a maximum level was reached. By contrast, Morandi et al.5 employed 0.42 mol of α-bromoisobutyryl bromide (BiB) to 1 g nanocrystals and converted 70% of the available surface hydroxyl groups after 24 h at 70 °C. They also used 16 or 24 h of immobilization time, when employing 0.21 mol of BiB, obtaining 25 and 35% conversion, respectively. As a suggestion, shorter immobilization times combined with an excess of the initiator may show a more pronounced time dependence.
To conclude, an initiator with a cleavable linker was successfully synthesized and immobilized on a cellulose surface, followed by polymerization of MMA in a controlled fashion via ARGET ATRP. The PMMA grafts were subsequently cleaved off from the surface, in the described non-destructive manner, characterized, and compared with the free polymer formed from the sacrificial initiator. From these results, it can be concluded that the surface and the bulk polymerization occur with similar kinetics, showing that the utilization of a sacrificial initiator is indeed a pragmatic approach to tailor the grafts. Furthermore, the initiator content was effectively estimated by employing Ellman's reagent, demonstrating that the immobilization of the initiator to the surface is a fast reaction. Noteworthy, the approach described in this paper is a versatile method suitable for a range of different substrates and polymers with various chemical handles, keeping in mind that the initiating moieties can be readily exchanged by other functionalities. Nevertheless, owing to that the initiator content on surfaces can be evaluated, post-functionalization reactions are significantly facilitated.
Finally, the Swedish research council is acknowledged for financial support. Anna Andersson is thanked for help with parts of the laboratory work.
Notes and references
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS.
- A. Carlmark and E. Malmström, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef CAS.
- M. Husseman, E. E. Malmström, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424–1431 CrossRef.
- J. Lindqvist and E. Malmström, J. Appl. Polym. Sci., 2006, 100, 4155–4162 CrossRef CAS.
- G. Morandi, L. Heath and W. Thielemans, Langmuir, 2009, 25, 8280–8286 CrossRef CAS.
- D. Nyström, J. Lindqvist, E. Östmark, A. Hult and E. Malmström, Chem. Commun., 2006, 3594–3596 RSC.
- D. Plackett, K. Jankova, H. Egsgaard and S. Hvilsted, Biomacromolecules, 2005, 6, 2474–2484 CrossRef CAS.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 2001, 123, 7497–7505 CrossRef CAS.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39–45 CrossRef CAS.
- S. Hansson, E. Östmark, A. Carlmark and E. Malmström, ACS Appl. Mater. Interfaces, 2009, 1, 2651–2659 CrossRef CAS.
- J. O. Zoppe, Y. Habibi, O. J. Rojas, R. A. Venditti, L.-S. Johansson, K. Efimenko, M. Österberg and J. Laine, Biomacromolecules, 2010, 11, 2683–2691 CrossRef CAS.
- D. Roy, J. T. Guthrie and S.b. Perrier, Macromolecules, 2005, 38, 10363–10372 CrossRef CAS.
- M. Barsbay, O. Gueven, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and L. Barner, Macromolecules, 2007, 40, 7140–7147 CrossRef CAS.
- M. Barsbay, O. Gueven, T. P. Davis, C. Barner-Kowollik and L. Barner, Polymer, 2009, 50, 973–982 CrossRef CAS.
- S. B. Lee, R. R. Koepsel, S. W. Morley, K. Matyjaszewski, Y. Sun and A. J. Russell, Biomacromolecules, 2004, 5, 877–882 CrossRef CAS.
- Q. Zhou, L. Greffe, M. J. Baumann, E. Malmström, T. T. Teeri and H. Brumer, III, Macromolecules, 2005, 38, 3547–3549 CrossRef CAS.
- J. Pyun, S. Jia, T. Kowalewski, G. D. Patterson and K. Matyjaszewski, Macromolecules, 2003, 36, 5094–5104 CrossRef CAS.
- H. Gao, N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 5995–6004 CrossRef CAS.
- J. Kamada, K. Koynov, C. Corten, A. Juhari, J. A. Yoon, M. W. Urban, A. C. Balazs and K. Matyjaszewski, Macromolecules, 2010, 43, 4133–4139 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087–3092 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1958, 74, 443–450 CrossRef CAS.
- C. Nilsson, E. Malmström, M. Johansson and S. M. Trey, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5815–5826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental information and results from FT-IR and UV analyses. See DOI: 10.1039/c0py00388c |
| This journal is © The Royal Society of Chemistry 2011 |