DOI:
10.1039/C0PY00385A
(Paper)
Polym. Chem., 2011,
2, 659-664
Bulk ring-opening polymerization (ROP) of L-lactide catalyzed by Ni(II) and Ni(II)-Sm(III) complexes based on a salen-type schiff-base ligand†
Received
24th November 2010
, Accepted 25th November 2010
First published on 22nd December 2010
Abstract
A monometallic (Ni2+, 1) and a bimetallic (Ni2+-Sm3+, 2) complexes based on the Salen-type Schiff-base ligand H22L (H22L = N,N′-bis(3-methoxysalicylidene)ethylene-1,2-diamine) were synthesized and characterized by FT-IR, ESI-MS and X-ray crystallography. The catalysis results showed that the two complexes with different active species, could efficiently catalyze the bulk solvent-free melt ring-opening polymerization (ROP) of L-lactide with moderate molecular weights and narrow molecular weight distributions. Especially, for the bimetallic complex 2, the involvement of rare ions effectively passivated the catalytic behavior on the ROP of L-lactide, while was in favor of the increase of polymeric molecular weights (Mn) and the polymerization controllability.
1 Introduction
Polylactide (PLLA) polymers, as one of the biodegradable, biocompatible polyesters for wide-ranging use in medicine or pharmaceutical related applications,1–3 are mostly synthesized by the ring-opening polymerization (ROP) of lactide using diverse catalysts,4 both in solution polymerization and in bulk solvent-free melt polymerization. In spite of the well-established achievements in solution polymerization,5 commercially, the ROP of lactide is most commonly carried out bybulk solvent-free melt polymerization, which exhibits advantages over solution polymerization: (i) no solvent is required; (ii) it is less vulnerable to impurity levels and unwanted side reactions; (iii) it is often useful for the large-scale production of PLLAs.6 From the viewpoint of needed catalysts, two kinds of catalysts have been reported: one is the metal complexes containing the characteristic initiators (alkoxide or carboxylate), and the mechanism of the ROP of lactide is the same as that in solution polymerization, and generally recognized to proceed via the reaction pathway shown in Scheme 1, involving the attack of a characteristic group on the ketonic group of a coordinated lactide molecule;7 the other is metal complexes lacking those normal initiators, while the excellent control of the polymerization could be well achieved from the molecular design of diverse catalysts and the selectivity of reaction conditions.8–14 In spite of the limited knowledge of the possible polymerization mechanism,15,16 the coordination geometry and Lewis acidity of active species are probably responsible for metal-mediated ROP of lactide. Although these impressive recent developments have been achieved, there has been an intense search for new generation catalysts for the bulk solvent-free melt ROP of lactide, especially bimolecular catalysts with Lewis acid centers and nucleophile centers.
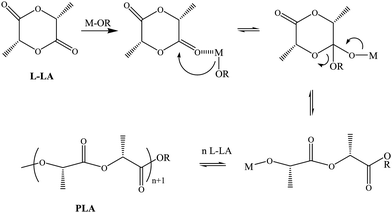 |
| | Scheme 1 Schematic representation of the ROP process of L-lactide initiated by the M-OR (OR, the characteristic initiators) complex. | |
Compared to the homogeneous or heterogeneous catalysis of Salen-type Schiff-base metal complexes in various chemical reactions,17–20 reports of their use for the ring-opening polymerization of lactide are relatively limited.21 Although many transition metal22–26 or lanthanide27 complexes of Salen-type chiral or achiral Schiff-base ligands are well known to be efficient catalysts for the ring-opening polymerization of L-lactide, they are limited to the solution polymerization. As for the bulk solvent-free melt ring-opening polymerization of L-lactide, many transition metal complexes, Cu2+ or Ni2+ complexes of phenoxy-ketimine ligands,12 and Ag+ or Au+ carbene complexes,9–11 have been reported to be active, in spite of lacking the normal characteristic initiators as used in solution polymerization. Especially, the recent report of bulk polymerization of L-lactide by Fe2+ or Mn3+Salen-type Schiff-base complexes revealed that they efficiently catalyzed the ring-opening polymerization of L-lactide at elevated temperatures under solvent-free melt conditions,28 while the structures of the catalysts are not clear, and to the best of our knowledge, no report of their mixed metal complexes for the bulk polymerization of L-lactide has been documented. Herein, we describe the syntheses, structures and the bulk solvent-free melt ROP of L-lactide of a monometallic (Ni2+, 1) and a bimetallic (Ni2+–Sm3+, 2) complexes based on the Salen-type Schiff-base ligand H22L (H22L = N,N′-bis(3-methoxysalicylidene)ethylene-1,2-diamine). The catalysis results showed that the two complexes (1–2) with different reactive centers could efficiently catalyze the polymerization of L-lactide with moderate molecular weights and narrow molecular weight distributions.
2 Experimental
All chemicals of reagent grade were commercially available and used without further purification. Element analyses were performed on a Perkin-Elmer 240C element analyzer. Infrared spectra were recorded on a Bruker EQUINOX55 FT-IR spectrophotometer in the region 4000–400 cm−1 in KBr pellets. ESI-MS was performed on a Finnigan LCQDECAXP HPLC-MSnmass spectrometer with a mass to charge (m/z) range of 2000 using a standard electrospray ion source and toluene as solvent. 1H NMR spectra were measured on a Varian Unity INOVA 400NB instrument using CDCl3 as solvent and TMS as internal standard at room temperature. Electronic absorption spectra in the UV-Visible region were recorded with a Hewlett Packard 8453 UV/Vis spectrophotometer. Thermogravimetric analyses were carried out on a NETZSCH TG 209 Instrument under flowing nitrogen by heating the samples from 25 to 600 °C. The GPC analyses of the molecular weight and molecular weight distributions (MWDs) of the polymers were performed on a Shimadzu LC-20AD system with tetrahydrofuran as the eluent at 40 °C with standard polystyrene as the reference. Styragel HMW 6E column (7.8 × 300 nm) was used. MALDI-TOF MS analysis was carried out on a commercial Reflux III MALDI-TOF mass spectrometer (Bruker Co., Germany) equipped with delayed extraction technology. Ions formed by a pulsed UV laser beam with 3 nm pulse (nitrogen laser, λ = 337 nm) were accelerated through 20 kV and detection voltage was set at 1.60 kV. The laser was adjusted by the experiments slightly above the threehold, and the mass spectra were obtained from the results of 35 laser shots in positive mode.
2.1 Syntheses of the Salen-type Schiff-base ligand H22L
The ligand H22L was synthesized by the typical procedure29,30 by condensation of o-vanillin (6.3 g, 40 mmol) and 1,2-diaminoethane (1.4 ml, 20 mmol) in absolute EtOH under reflux for about 5 h. After cooling to room temperature, the insoluble precipitate was filtered and re-crystallized using absolute EtOH to give the yellow polycrystalline solid. Yield: 5.0 g, 76%. Calc. for C18H20N2O4: C 65.84, H 6.14, N 8.53%; Found: C, 65.58, H, 6.06, N, 8.63%; IR (KBr, cm−1): 3444 (b), 3001 (w), 2930 (w), 2840 (w), 1632 (s), 1468 (s), 1410 (m), 1249 (s), 1080 (m), 958 (m), 736 (m), 695 (w), 642 (w), 564 (w), 525 (w). 1H NMR (400 MHz, CDCl3): δ, 13.52 (s, 2H, –OH), 8.55 (s, 2H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 6.98 (m, 4H, –Ph), 6.75(t, 2H, Ph), 3.91 (s, 4H, –CH2), 3.74 (s, 6H, –MeO).
N), 6.98 (m, 4H, –Ph), 6.75(t, 2H, Ph), 3.91 (s, 4H, –CH2), 3.74 (s, 6H, –MeO).
2.2 Synthesis of [NiL] (1)
To a stirred solution of H22L (0.164 g, 0.5 mmol) in absolute CH2Cl2 (5 ml), Ni(OAc)2·4H2O (0.125 g, 0.5 mmol) was added and heated under reflux for 5 h. The mixture was allowed to cool to room temperature and filtered. The resultant clear brown solution was left to stand at room temperature for several days to give green polycrystalline product of 1 in 70% yield. Calc. for C18H18N2O4Ni: C, 56.15; H, 4.71; N, 7.28%; found: C, 56.38; H, 4.66; N, 7.35%. IR (KBr, cm−1): 3291 (b), 2923 (w), 2828 (w), 1646 (s), 1603 (w), 1444 (vs), 1240 (m), 1215 (s), 728 (m), 697 (w), 636 (w), 582 (w), 531 (w), 463 (w). 1H NMR (400 MHz, CDCl3): δ, 8.51(s, 2H, –CHN), 7.05 (m, 4H, –Ph), 6.55 (t, 2H, Ph), 3.80 (s, 4H, –CH2), 3.74 (s, 6H, –MeO). ESI-MS (m/z): 386 [M–H]+.
2.3 Synthesis of [Ni(L)Sm(NO3)3] (2)
To a solution of 1 (0.116 g, 0.3 mmol) in absolute EtOH (4 ml), a solution of Sm(NO3)3·6H2O (0.134 g, 0.3 mmol) in absolute CH3COCH3 (5 ml) was added, and the mixture was refluxed for 3 h. Then 1 ml absolute DMF was added to give a clear brown solution. Diethyl ether was allowed to diffuse slowly into this solution at room temperature and deep red polycrystalline product of 2 was obtained in a few weeks. Yield: 0.147 g (68%). Calc. for C18H18N5O13NiSm: C, 29.97; H, 2.51; N, 9.71%; found: C, 29.92; H, 2.72; N, 9.66%. IR (KBr, cm−1): 3424 (w), 2987 (w), 2846 (w), 1640 (s), 1561 (w), 1498 (s), 1466 (vs), 1316 (s), 1276 (s), 1235 (s), 1166 (m), 1109 (w), 1080 (m), 1027 (w), 989 (w), 956 (w), 861 (w), 811 (w), 784 (w), 739 (m), 685 (w), 625 (w), 582 (w), 495 (w). ESI-MS (m/z): 659 [M–NO3]+.
2.4 Structure determination
Single crystal of [Ni(L)Sm(NO3)3]·CH3COCH3 (2·CH3COCH3), of suitable dimensions was mounted onto glass fibers for crystallographic analyses. For 2·CH3COCH3, all the intensity data were collected at 293(2) K on a Bruker SMART CCD diffractometer (Mo-Kα radiation, λ = 0.71073 Å) in Φ and ω scan modes. Structure was solved by direct methods followed by difference Fourier syntheses, and refined by full-matrix least-squares techniques against F2 using SHELXTL.31 All the non-hydrogen atoms were refined with anisotropic thermal parameters. Absorption corrections were applied using SADABS.32 All hydrogen atoms were placed in calculated positions and refined isotropically using a riding model. Crystallographic data and refinement parameters for the complex 2·CH3COCH3 are presented in Table 1. Selected bond distances and bond angles for 2·CH3COCH3 are given in Table 2.
| Compound |
2·CH3COCH3 |
| Empirical formula |
C21H24NiN5O14Sm
|
| Formula weight |
779.51 |
| Crystal system |
Monoclinic |
| Space group |
Cc
|
|
a/Å |
11.276(2) |
|
b/Å |
26.738(5) |
|
c/, Å |
9.177(2) |
|
α (°) |
94.950(5) |
|
β (°) |
90 |
|
γ (°) |
94.07(3) |
|
V/Å3 |
90 |
|
Z
|
4 |
|
ρ/g cm−3 |
1.876 |
|
T/K |
293(2) |
| Crystal size/mm |
0.33 × 0.26 × 0.23 |
|
μ/mm−1 |
2.867 |
|
F(000) |
1548 |
| Data/restraints/parameters |
3525/2/380 |
| Quality-of-fit indicator |
0.827 |
| No. Unique reflections |
3525 |
| No. Observed reflections |
7443 |
| [I > 2σ(I)] |
|
|
R
|
0.0520 |
|
W
R
|
0.1147 |
Table 2 Interatomic distances (Å) and bond angles (°) with esds for 2·CH3COCH3
|
2·CH3COCH3 |
| Ni(1)–N(1) |
1.790(20) |
Ni(1)–N(2) |
1.824(17) |
| Ni(1)–O(2) |
1.846(12) |
Ni(1)–O(3) |
1.844(12) |
| Sm(1)–O(1) |
2.625(13) |
Sm(1)–O(2) |
2.418(12) |
| Sm(1)–O(3) |
2.366(12) |
Sm(1)–O(4) |
2.556(14) |
| Sm(1)–O(5) |
2.484(14) |
Sm(1)–O(7) |
2.330(30) |
| Sm(1)–O(8) |
2.543(16) |
Sm(1)–O(10) |
2.408(18) |
| Sm(1)–O(11) |
2.494(12) |
Sm(1)–O(13) |
2.540(30) |
| N(1)–Cu(1)–N(2) |
81.1(11) |
N(1)–Cu(1)–O(2) |
98.4(9) |
| N(1)–Cu(1)–O(3) |
179.7(9) |
O(1)–Sm(1)–O(4) |
153.9(5) |
| O(2)–Sm(1)–O(3) |
60.7(4) |
O(5)–Sm(1)–O(7) |
49.6(6) |
| O(8)–Sm(1)–O(10) |
50.5(5) |
O(11)–Sm(1)–O(13) |
50.2(7) |
L-lactide was prepared from L-lactic acid as previously reported.33 The crude product was further purified by re-crystallization three times from dried ethyl acetate, then dried for 24 h in vacuum at 30 °C. Under nitrogen, 1.000 g (6.94 mmmol) of the freshly re-crystallized L-lactide monomer and the catalyst 1 or 2, in a stipulated molar ratio ([M]/[C]), were charged in an ampoule inside a glove box. The ampoule was put under high vacuum (about 12 Pa) for one hour, after which, the ampoule was sealed under vacuum. The polymerizations were performed in a thermostatically controlled oil bath at 130 or 160 °C for the selected time. Subsequently, the molten reactive polymer mixture was cooled by immersing the sealed ampoule in liquid nitrogen and terminated by introducing absolute MeOH with 5% (w/w) HCl to stop the polymerization. The resulting polylactide polymer was dissolved in absolute acetone and precipitated in water. The filtered precipitate was dried under vacuum until a constant weight was reached. Molecular weights (Mw and Mn) and molecular weight distributions (PDI = Mw/Mn) were determined by gel permeation chromatography (GPC) with polystyrene as standard.
3 Results and discussion
3.1 Synthesis and characterization
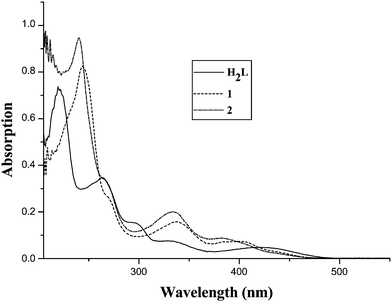
As shown in Scheme 2, reaction of an equimolar amount of H22L and Ni(OAc)2·4H2O in absolute EtOH afforded complex 1 in good yield. Further reaction of 1 with Sm(NO3)3·6H2O in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in EtOH–DMF resulted in the formation of complex 2 in a good yield of ca. 68%. The IR spectra of 1–2 show the characteristic absorptions of the ν(CN) vibration at 1642 cm−1 for 1 and 1640 cm−1 for 2, are slightly blue shifted by ca. 8 cm−1 relative to those of the free salen-type ligand H22L (1632 cm−1 for H22L) upon coordination of metal ions.34 For 2, two strong absorption bands at 1466 and 1316 cm−1 attributed to ν(NO3−).34Thermogravimetric analysis (TGA) in the range 25 to 600 °C showed that the framework of 1 or 2 decomposes at temperatures over 298 °C (over the selected polymerization temperatures) where an abrupt weight loss was observed, followed by a final weight loss in the 305 (1) or 313 (2) to 600 °C range. As to the solution behavior of complexes 1–2, the ESI-MS spectra exhibit one peak at m/z 386 and 659, respectively, corresponding to the major species [NiL]+ and [NiLSm(NO3)2]+, and indicating that the respective discrete monometallic or bimetallic molecule exists in dilute CHCl3 solution. Further from the UV-Visible absorption spectra of the ligand and the two complexes examined in CHCl3 solution, as shown in Fig. 1, the free ligand H22L exhibits absorption bands at 220, 264, 296, 334 and 429 nm, which could be assigned to the ligand-centered π → π* transitions, while for the two complexes 1–2, the lower energy bands are significantly blue-shifted upon coordination to metal ions (243, 338 and 400 nm for 1; 241, 334 and 385 nm for 2).
:1 molar ratio in EtOH–DMF resulted in the formation of complex 2 in a good yield of ca. 68%. The IR spectra of 1–2 show the characteristic absorptions of the ν(CN) vibration at 1642 cm−1 for 1 and 1640 cm−1 for 2, are slightly blue shifted by ca. 8 cm−1 relative to those of the free salen-type ligand H22L (1632 cm−1 for H22L) upon coordination of metal ions.34 For 2, two strong absorption bands at 1466 and 1316 cm−1 attributed to ν(NO3−).34Thermogravimetric analysis (TGA) in the range 25 to 600 °C showed that the framework of 1 or 2 decomposes at temperatures over 298 °C (over the selected polymerization temperatures) where an abrupt weight loss was observed, followed by a final weight loss in the 305 (1) or 313 (2) to 600 °C range. As to the solution behavior of complexes 1–2, the ESI-MS spectra exhibit one peak at m/z 386 and 659, respectively, corresponding to the major species [NiL]+ and [NiLSm(NO3)2]+, and indicating that the respective discrete monometallic or bimetallic molecule exists in dilute CHCl3 solution. Further from the UV-Visible absorption spectra of the ligand and the two complexes examined in CHCl3 solution, as shown in Fig. 1, the free ligand H22L exhibits absorption bands at 220, 264, 296, 334 and 429 nm, which could be assigned to the ligand-centered π → π* transitions, while for the two complexes 1–2, the lower energy bands are significantly blue-shifted upon coordination to metal ions (243, 338 and 400 nm for 1; 241, 334 and 385 nm for 2).
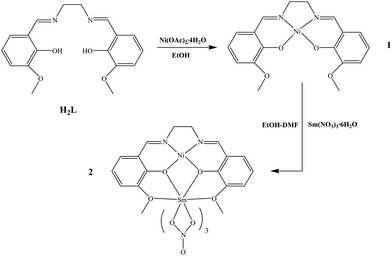 |
| | Scheme 2 Reaction scheme for the syntheses of complexes 1–2 | |
3.2 Structures
For complex 1, the room temperature 1H NMR spectrum in CDCl3 exhibits one set of the proton resonances of the L2−2− ligand, significantly red-shifted in relative to those of the free ligand, which shows a typical monometallic structure of [NiL] in solution. The Ni complex 1 and its analogous structures have been previously reported by some groups.35–37 The X-ray crystal structure analysis of the complex [NiLSm(NO3)3]·CH3COCH3 (2·CH3COCH3) unambiguously revealed the bimetallic Ni-Sm structure, as depicted in Fig. 2. The neutral 2·CH3COCH3 revealed that the relatively soft Ni2+ ion is located in the inner N2O2 core, the relatively hard Sm3+ ion in the outer O2O2 cavity of the Schiff base L2−2− ligand. The Ni2+ ion lies in a four-coordinate environment and adopts a slightly distorted square-planar geometry, with two sets of unequal Ni–N(imino) (1.790(20)–1.824(17) Å) and Ni–O (phenolic) (1.844(12)–1.846(12) Å) bond lengths and N–Ni–O bond angles (98.4(9)–98.6(10)° and 178.8(7)–179.7(9)°). The Sm3+ ion is ten coordinated and bound by four O atoms from the Schiff base L2−2− ligand, and six from three bidentate NO3−groups. The Sm–O bond lengths depend on the nature of the oxygen atoms: they vary from 2.330(30) to 2.625(13) Å, and the bond lengths from phenoxo oxygen atoms are shorter that those from oxygen atoms of NO3− anions. It is worth noting that the coordination geometry of the metal ions on the Schiff base L2−2− ligand in 2·CH3COCH3 is comparable to those of magnetic Cu–Ln38 while incomparable to those of luminescent Zn–Ln39Salen-type Schiff-base complexes. The solvates are not bound to the framework and they exhibit no observed interactions with the host structure.
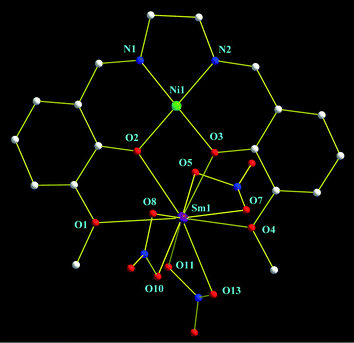 |
| | Fig. 2 Structure scheme of complex 2, Hydrogen atoms and solvates are omitted for clarity. | |
3.3 The polymerization studies of L-lactide
The ROP of L-lactide using complexes 1–2 as catalysts in a stipulated molar ratio of [M]/[C] were carried out at 130 or 160 °C for the selected time under solvent-free melt conditions. Under these conditions (entry 5–16), the reaction mixture would form a monomer melt in which the polymerization would occur, and the results were summed in Table 3. For complex 1, at the polymerization temperature of 130 °C, the catalytic activities increased with the increase of reaction times, and the well-controlled polymerization was observed from the narrow molecular weight distributions (PDI = 1.17–1.39). The highest Mn and the smallest PDI value (PDI = 1.17) of the products were achieved by polymerization for 24 h and the molecular weight distribution (PDI = 1.39) broadened with prolonged polymerization time, which should be attributed to the thermal depolymerization.40 Similar to the polymerization for the prolonged time, the result of ROP of L-lactide performed at temperatures ranging from 130 to 160 °C for 24 h showed that with increasing temperature, the monomer conversion increased, however, the molecular weight decreased and the molecular weight distribution broadened. This may also be ascribed to the thermal depolymerization and the acceleration of transesterification as the polymerization temperature increases. When the [M]/[C] ratio was lower than [1000]/[1], little difference was found in spite of the increase of the polymer yields and the catalytic activities. The molecular weights (Mn and Mw) of the polymerization products were also affected by the [M]/[C] ratio: The maximum of molecular weight (Mn or Mw) was achieved at an [M]/[C] ratio of [2000]/[1], which showed that the lower catalyst concentration could be helpful for the growth of the polymeric products; While a lower catalyst concentration will produce less initiation sites, thus leading to the lower catalytic activities.41 It is worth noting that in the selected range ([500–2000]/[1]) of [M]/[C] ratios, the Ni2+ catalyst 1 could afford the PLAs with narrow molecular weight distributions (Mw/Mn = 1.17–1.24), while the blank experiment (entry 1), the presence of free ligand or inorganic salt (entry 2 or entries 3–4) showed that no polymeric products were obtained, both further demonstrates that the presence of single active species (Ni2+) in 1 endows the controllable polymerization process.42
Table 3
Catalytic data in the bulk polymerization of L-lactide
| Entry |
Compound |
Time/h |
T/°C |
M/Ca ratio |
Conversionb (%) |
Activity/g mol−1 h−1 |
M
n
c/103 |
M
w
d/103 |
PDIe |
|
[M]/[C] ratio is the molar ratio of L-lactide monomer and catalyst.
Conversion is determined by 1H NMR spectroscopy.
Mn is the relative number-average molecular weight.
Mw is the relative weight-average molecular weight.
PDI = Mw/Mn.
No polymeric product was obtained.
|
| 1 |
— |
24 |
130 |
|
— |
0 |
—f |
— |
— |
| 2 |
Ligand |
24 |
130 |
1000/1 |
— |
0 |
— |
— |
— |
| 3 |
Ni(OAc)2 |
24 |
130 |
1000/1 |
— |
0 |
— |
— |
— |
| 4 |
Sm(NO3)3 |
24 |
130 |
1000/1 |
— |
0 |
— |
— |
— |
| 5 |
1
|
12 |
130 |
1000/1 |
61 |
240.5 |
10.383 |
12.228 |
1.18 |
| 6 |
1
|
24 |
130 |
1000/1 |
69 |
392.2 |
22.702 |
26.635 |
1.17 |
| 7 |
1
|
36 |
130 |
1000/1 |
71 |
398.4 |
22.596 |
31.385 |
1.39 |
| 8 |
1
|
24 |
160 |
1000/1 |
83 |
402.7 |
20.829 |
31.994 |
1.54 |
| 9 |
1
|
24 |
130 |
500/1 |
72 |
401.0 |
22.071 |
26.662 |
1.21 |
| 10 |
1
|
24 |
130 |
2000/1 |
52 |
110.7 |
25.494 |
31.664 |
1.24 |
| 11 |
2
|
12 |
130 |
1000/1 |
55 |
289.3 |
17.381 |
20.235 |
1.16 |
| 12 |
2
|
24 |
130 |
1000/1 |
65 |
375.3 |
26.940 |
30.146 |
1.12 |
| 13 |
2
|
36 |
130 |
1000/1 |
68 |
384.6 |
26.705 |
30.791 |
1.15 |
| 14 |
2
|
24 |
160 |
1000/1 |
70 |
398.8 |
24.045 |
29.431 |
1.22 |
| 15 |
2
|
24 |
130 |
500/1 |
76 |
419.4 |
22.389 |
29.531 |
1.32 |
| 16 |
2
|
24 |
130 |
2000/1 |
56 |
180.8 |
25.883 |
34.425 |
1.33 |
For complex 2, on the varied reaction conditions, the better controllable polymerization of L-lactide was observed, which was also summed in Table 3. It is significant on the correlation on the molecular structure of complex 2versus the catalytic behavior. The further coordination of Sm(NO3)3 in complex 2 endowed the slight distortion of square-planar geometry and the increase of Lewis acidity of the four-coordinate Ni2+ ion. On the condition of short reaction time (12 h) or high catalyst concentration ([M]/[C] = [500]/[1]) at 130 °C, complex 2 showed higher catalytic activity than that of complex 1, respectively. When the reaction time was prolonged or the catalyst concentration was increased, an slight decrease of catalytic activities was observed, which shows that the steric effect from the coordination of Sm(NO3)3 in complex 2 should prevent the coordination with the active species (Ni2+ ion) for L-lactide monomers. At the same time, due to the existence of nitrates around the Sm3+ ions in complex 2, in agreement with its solution behavior, the second metal center (Sm3+) was inactive to the ROP of L-lactide. So in spite of the design of bimolecular structure, complex 2 has a typical monomolecular active center, like that of complex 1. However, for the polymeric products obtained by complex 2 (entries 12–14), a significant increase in molecular weights (Mn) and a lower PDI (Mw/Mn) value were observed, which suggests that the steric effect from the coordinated Sm(NO3)3 in complex 2 is much more suitable for the growth of polymer chains by further insertion for L-lactide monomers with good polymerization controllability, even at 160 °C for 24 h or 130 °C for 36 h. It is noteworthy that, in contrast to other known catalysts with characteristic initiators for the ring-opening polymerization of L-lactide in coordination-insertion mechanism,5,22–27 complexes 1–2 without those initiators, also efficiently catalyze the bulk solvent-free melt ring-opening polymerization (ROP) of L-lactide with moderate catalytic activities (as shown in Table 3), as found for the Fe2+ or Mn3+Salen-type Schiff-base ligands.28 Moreover, based on the further investigation at elevated temperatures and prolonged reaction times with the catalysts 1–2, the involvement of Sm3+ ion in 2, endows the feasibility to obtain the PLLA product in the controllable mode, even on the condition of 160 °C for 24 h or 130 °C for 36 h, compared to that of 1 at low temperature (130 °C) and short reaction time (24 h).
Thermogravimetric analysis (TGA) in the range 25–600 °C showed that these selected polymeric products (entry 6 and entry 12) decomposed at temperatures over 240 °C (243 °C for 1 and 261 °C for 2), where an abrupt weight loss was observed, followed by a final weight loss in the 375–600 °C range. The decomposed temperature is relative to the length of the polymeric chain, as proved by the larger Mn or Mw for the polymerization from 2. One of the identical 1H NMR spectra of the polymers (as shown in Fig. 3) showed a doublet at 1.59 ppm for the methyl (cH) and a multiplet at 5.18 ppm for the methine (bH) of main chain of PLA, indicating no inversion of the configuration of asymmetric carbon atoms of the monomer under the reaction conditions. The additional signals appearing at 1.49, 3.04 and 4.36 ppm should be assigned to the methyl, the hydroxyl end group and the methine of PLLA, respectively.43 It is worth noting that the systems 1–2 are active for the bulk ROP of L-Lactide in the absence of normal initiators (alkoxide, amide or carboxylate), and the end groups of PLLA are not from the catalysts, as also checked by the 1H NMR spectroscopy (as shown in Fig. 3) and the MALDI-TOF results (as shown in the ESI†), but probably from the replacement of methyl groups in either the impurity (alcohol or lactic acidetc.), the monomer or alcohol when terminating the polymerization with methanol containing 5% HCl. Thus, the initiation may occur through the metal-alkoxide bond of the catalysts, through the stable and selective acyl-oxygen bond cleavage of the monomer, and the ring-opening polymerization also maybe proceeds via the typical “coordination-insertion” mechanism.44
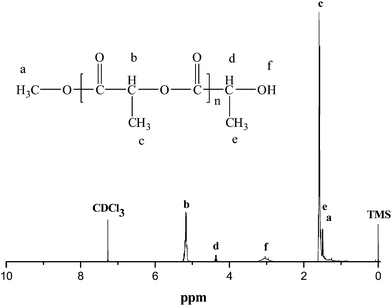 |
| | Fig. 3
1H NMR spectrum in CDCl3 of PLA prepared by complex 1 or 2 from ROP of L-lactide. | |
In conclusion, the monometallic Ni2+ complex 1 and the bimetallic Ni2+-Sm3+ complex 2 were shown to efficiently catalyze the bulk solvent-free melt ring-opening polymerization (ROP) of L-lactide with moderate molecular weights and narrow molecular weight distributions. The correlation of molecular structure versuscatalytic activity showed that the different catalytic behaviors resulted from the different active species, especially, the involvement of rare ions slightly passivated the catalytic behaviors on the ROP of L-lactide, being in favor of the increase of polymeric molecular weights (Mn) and the polymerization controllability. With this in mind, the design of more active and multi-molecular active species catalysts is now under way.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (CUGL100407) and the National Natural Science Foundation of China (50903077, 20871098), the Natural Science Foundation of Hubei Province (2009CDB163, 2008CDZ065), the State Key Laboratory of Structural Chemistry (20100014), the Provincial Key Item of Shaanxi, Graduate Innovation and Creativity Funds (08YZZ48), Graduate Cross-discipline Funds (09YJC23) and Bachelor Graduate Innovation and Creativity Funds of Northwest University in P. R. of China.
References
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef
 .
.
- M. Vert, Biomacromolecules, 2005, 6, 538 CrossRef CAS .
- J. H. Khan, F. Schue and G. A. George, Polym. Int., 2009, 58, 296 CrossRef CAS .
- J. C. Wu, T. L. Yu, C. T. Chen and C. C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS .
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11 Search PubMed .
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS .
- A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahan, Angew. Chem., Int. Ed., 2007, 46, 2280 CrossRef CAS .
- X. Y. Wang, K. R. Liao, D. P. Quan and Q. Wu, Macromolecules, 2005, 38, 4611 CrossRef CAS .
- M. K. Samantaray, V. Katiyar, D. Roy, K. Pang, H. Nanavati, R. Stephen, R. B. Sunoj and P. Ghosh, Eur. J. Inorg. Chem., 2006, 2975 CrossRef CAS .
- L. Ray, V. Katiyar, M. J. Raihan, H. Nanavati, M. M. Shaikh and P. Ghosh, Eur. J. Inorg. Chem., 2006, 3724 CrossRef CAS .
- L. Ray, V. Katiyar, S. Barman, M. J. Raihan, H. Nanavati, M. M. Shaikh and P. Ghosh, J. Organomet. Chem., 2007, 692, 4259 CrossRef CAS .
- A. John, V. Katiyar, K. Pang, M. M. Shaikh, H. Nanavati and P. Ghosh, Polyhedron, 2007, 26, 4033 CrossRef CAS .
- A. D. Schwarz, A. L. Thompson and P. Mountford, Inorg. Chem., 2009, 48, 10442 CrossRef CAS .
- J. Börner, U. Flörke, T. Glöge, T. Bannenberg, M. Tamm, M. D. Jones, A. Döring, D. Kuckling, S. Herres-Pawlis and J. Mole, J. Mol. Catal. A: Chem., 2010, 316, 139 CrossRef .
- E. L. Marshall, V. C. Gibson, H. S. Rzepa and J. Am, J. Am. Chem. Soc., 2005, 127, 6048 CrossRef CAS .
- M. H. Chisholm, N. J. Patmore and Z. P. Zhou, Chem. Commun., 2005, 127 RSC .
- N. S. Venkataramanan, S. Natarajan, G. Kuppuraj and S. Rajagopai, Coord. Chem. Rev., 2005, 249, 1249 CrossRef CAS .
- C. Baleizao and H. Garcia, Chem. Rev., 2006, 106, 3987 CrossRef CAS .
- R. Ma, Y. B. Hou, J. Gao and F. Bao, Polym. Rev., 2009, 49, 249 Search PubMed .
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420 CrossRef CAS .
- D. A. Atwood and M. J. Harvey, Chem. Rev., 2001, 101, 37 CrossRef CAS .
- P. A. Cameron, D. Jhurry, V. C. Gibson, A. J. P. White, D. J. Williams and S. Williams, Macromol. Rapid Commun., 1999, 20, 616 CrossRef CAS .
- J. C. Wu, B. H. Huang, M. L. Hsueh, S. L. Lai and C. C. Lin, Polymer, 2005, 46, 9784 CrossRef CAS .
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343 CrossRef CAS .
- R. J. C. Atlinson, K. Gerry, V. C. Gibson, N. J. Long, E. L. Marshall and L. J. West, Organometallics, 2007, 26, 316 CrossRef CAS .
- M. H. Chisholm, J. C. Gallucci, K. T. Quisenberry and Z. P. Zhou, Inorg. Chem., 2008, 47, 2613 CrossRef CAS .
- E. Grunova, E. Kirillow, T. Roisnel and J. F. Carpentier, Organometallics, 2008, 27, 5691 CrossRef CAS .
- B. B. Idage, S. B. Idage, A. S. Kasegaonkar and R. V. Jadhav, Mater. Sci. Eng., B, 2010, 168, 193 CrossRef CAS .
- X. Q. Lü, W. Y. Bi, W. L. Chai, J. R. Song, J. X. Meng, W. Y. Wong, W. K. Wong and R. A. Richard, New J. Chem., 2008, 32, 127 RSC .
- X. Q. Lü, W. Y. Wong and W. K. Wong, Eur. J. Inorg. Chem., 2008, 523 .
-
G. M. Sheldrick, SHELXS-97: Program for Crystal Structure Refinement, Göttingen, Germany, 1997 Search PubMed .
-
G. M. Sheldrick, SADABS, University of Göttingen, 1996 Search PubMed .
- K. S. Anderson, K. M. Schreck and M. A. Hillmyer, Polym. Rev., 2008, 48, 85 Search PubMed .
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 4th edn, 1986, sect. II. 8 Search PubMed .
- W. W. Fee, J. D. Pulsford and P. D. Vowle, Aust. J. Chem., 1973, 26, 675 CAS .
- Z. Guo, L. Li, T. Xu, J. H. Li and D. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1158 CrossRef .
- Y. Y. Yu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m948 CrossRef .
- W. B. Sun, P. F. Yan, G. M. Li, J. W. Zhang and H. Xu, Inorg. Chim. Acta, 2009, 362, 1761 CrossRef CAS .
- W. Y. Bi, X. Q. Lü, W. L. Chai, W. J. Jin, J. R. Song and W. K. Wong, Inorg. Chem. Commun., 2008, 11, 1316 CrossRef CAS .
- S. H. Hyon, K. Jamshidi and Y. Ikada, Biomaterials, 1997, 18, 1503 CrossRef CAS .
- J. P. Puaux, I. Banu, I. Nagy and G. Bozga, Macromol. Symp., 2007, 259 .
- M. H. Chisholm and Z. P. Zhou, J. Mater. Chem., 2004, 14, 3081 RSC .
- X. B. Pan, A. Liu, X. Z. Yang, J. C. Wu and N. Tang, Inorg. Chem. Commun., 2010, 13, 376 CrossRef CAS .
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available: MALDI-TOF result analysis. CCDC reference numbers 776507. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0py00385a |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.