Photocrosslinking the polystyrene core of block-copolymer nanoparticles†
Siyan
Zhang
a,
Douglas H.
Adamson
c,
Robert K.
Prud'homme
*a and
A. James
Link
*ab
aDepartment of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA. E-mail: ajlink@princeton.edu; prudhomm@princeton.edu; Fax: +609-258-0211
bDepartment of Molecular Biology, Princeton University, Princeton, NJ 08544, USA
cDepartment of Chemistry and Institute of Materials Science, University of Connecticut, Storrs, CT 06269, USA
First published on 20th December 2010
Abstract
The crosslinking of the core of nanoparticles composed of polystyrene-block-poly(ethylene oxide) copolymers can be achieved through encapsulation of a small molecule aryl diazide, 4,4′-diazidobiphenyl, and subsequent photolysis. The core-crosslinked nanoparticles exhibited high stability under thermal challenge. These stabilized nanoparticles have potential to serve as a nanobead for the polymerase chain reaction (PCR). We also demonstrated that this crosslinker can endow thin films of polystyrene with solvent resistance. NMR studies on these films provided evidence that crosslinking was occurring viainsertion of the nitrene formed by photolysis of the azide into the methylene or methine groups in the backbone of polystyrene.
Introduction
Block copolymer nanoparticles hold promise in a number of fields including drug delivery and imaging.1–3 The ability to functionalize both the core4,5 and the surface6,7 of block-copolymer nanoparticles is a major advantage of these materials. We have previously described a method (Fig. 1), termed Flash NanoPrecipitation (FNP)8,9 to generate nanoparticles (NPs) ranging in diameter from 20–500 nm from amphiphilic block copolymers such as polystyrene–poly(ethylene oxide) (PS-b-PEO)10 and poly(caprolactone)–poly(ethylene oxide) (PCL-b-PEO).11 While FNP was initially developed in order to encapsulate hydrophobic payloads in the core of a nanoparticle, more recently we have turned our attention to surface functionalization of these particles.6 One avenue of this work in which we are particularly interested is developing polymeric nanoparticles with covalently attached single-stranded DNA. An application for such particles is as a replacement for larger, micron-scale latex beads used in some massively parallel signature sequencing (MPSS) efforts.12–15 In these applications of MPSS, a single DNA fragment is amplified via the polymerase chain reaction (PCR) such that many copies of a single sequence are present on a single bead. Millions of individual beads are simultaneously imaged to determine the DNA sequence. Being able to utilize smaller NPs with extensive surface functionalization with DNA could increase the throughput of MPSS substantially. One hurdle in using block-copolymer nanoparticles in MPSS is that the particles must be able to withstand the thermally demanding conditions of PCR, including heating to 95 °C to denature double stranded DNA as well as rapid thermal cycling between ∼50 °C and 95 °C. The use of a polymer with a high glass transition temperature (Tg), such as PS, can begin to satisfy the thermal demands of PCR, however, we observed that even NPs with PS cores were unstable during PCR thermal cycling. Here we describe an aromatic diazide crosslinker that efficiently stabilizes the core of PS-b-PEO block copolymer nanoparticles by photocrosslinking the PS core. While aromatic diazides have been used previously in photoresists16–18 including polystyrene,19 our work represents the first description, to our knowledge, of using these molecules to stabilize the core of nanoparticles.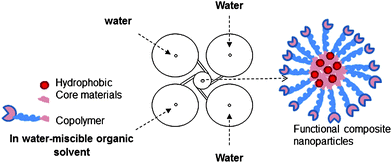 | ||
| Fig. 1 Schematic of Flash NanoPrecipitation (FNP). A water-miscible organic stream containing block-copolymer, homopolymer, and hydrophobic payload is rapidly combined with water streams in an impinging jet mixer. Kinetically trapped nanoparticles are formed in which the hydrophobic materials are sequestered in the core of the particle. | ||
In order to be a suitable small molecule crosslinker, the molecule must be inert before activation, reactive upon activation, and hydrophobic enough to be partitioned into the PS core of a PS-b-PEO nanoparticle (Fig. 2A). The most common method for generating crosslinked PS is to randomly copolymerize a bifunctional monomer, such as divinylbenzene.20 Previously 1,3-benzenedisulfonyl azide has been used as a thermally initiated PS crosslinker, but its use required that the PS be in a melt.21 Neither of these routes is compatible with block-copolymer NP formation. Peppas and Bussing used Friedel–Crafts chemistry to crosslink PS post-polymerization,22 but the requirement for organic solvent and elevated temperature makes this method incompatible with core–shell NPs as well. Photochemistry is an alternative route of activating the crosslinking agent that can be carried out at ambient temperature and under aqueous conditions. Given these considerations and previous demonstrations of their utility in crosslinking of PS photoresists, aryl azides appeared to be a promising photoactivatable moiety. When excited with UV light, aryl azides decompose to highly reactive nitrenes (Fig. 2B),23 which can then insert into aliphatic C–H bonds, such as the methylene and methine groups found in the polystyrene backbone. Here we have evaluated the effectiveness of aryl diazides as crosslinkers in nano-confined polystyrene core (Fig. 2A) of copolymer nanoparticles and performed NMR studies to characterize the chemical nature of the crosslinks formed.
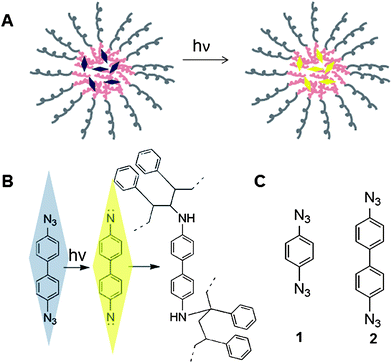 | ||
| Fig. 2 Photoinitiated aromatic diazide crosslinkers in the core of block-copolymer nanoparticles (NP). (A) Schematic of PS-b-PEO NP with UV-initiated crosslinker encapsulated in its core. (B) Conversion of aryl azide to reactive nitrene upon irradiation. The nitrene is capable of inserting into aliphatic C–H bonds such as the methylene group (top) or the methine group (bottom) found in the PS backbone. (C) Structure of crosslinkers synthesized in this study: 1, 1,4-diazidobenzene; 2, 4,4′-diazidobiphenyl. | ||
Experimental
Synthesis of aromatic diazides
General: all solvents and reagents were obtained from Sigma-Aldrich unless otherwise noted. Caution: since the aromatic diazides 1 and 2 (Fig. 2C) are potentially high energy materials, all reactions and separations were carried out at ambient temperature or below.1,4-Diazidobenzene (1) was synthesized by a known procedure with some modifications.24 In a flame-dried 250 mL round-bottom flask, 1,4-diaminobenzene (3.0 g, 27.8 mmol) was added to dry CH3CN (100 mL) in an ice bath. While stirring in an ice bath, tert-butyl nitrite (t-BuONO, 90% pure, 9.53 g, 83.3 mmol, 3 eq.) and trimethylsilyl azide (TMSN3, 95% pure, 8.07 g, 66.7 mmol, 2.4 eq.) were added dropwise sequentially. The resulting solution was allowed to warm up to room temperature and stirred for 20 h with the flask covered by aluminium foil to prevent light exposure. The reaction mixture was concentrated under vacuum at RT, and the crude product was suspended in hexane/ethyl acetate (1/1). The soluble fraction was purified by silica gel chromatography with hexane/ethyl acetate (1/1) to afford 1-azido-4-aminobenzene (1.8 g, 13.4 mmol, 48% yield) as amber crystals. 1-Azido-4-aminobenzene (0.5 g, 3.73 mmol) was subjected to a second round of azidation for 20 h (t-BuONO: 2.57 g, 22.4 mmol, 6 eq.; TMSN3: 2.71 g, 22.4 mmol, 6 eq.) to afford 1,4-diazidobenzene (0.48 g, 3.00 mmol, 80%) as amber crystals after flash chromatography on silica gel with hexane. Notably, while Miyake and Chujo reported that 1 could be obtained from only one cycle of azidation, we found that only one of the amines was converted to azide after 20 h of azidation. A second azidation reaction with fresh reactants was able to drive the reaction to the diazide. 1H NMR (500 MHz, CDCl3) δ: 7.01 (s, 4H).
4,4′-Diazidobiphenyl (2) was synthesized in a similar fashion as 1 from commercially available benzidine. Benzidine (95% pure, 1 g, 5.16 mmol), t-BuONO (4.09 mL, 31 mmol, 6 eq.) and TMSN3 (4.57 mL, 31 mM, 6 eq.) were reacted in 120 mL dry CH3CN for 2 d to afford 1,4-diazidobiphenyl (0.95 g, 78%) as amber crystals after chromatographic purification with hexane. In contrast to the synthesis of 1, only a single, longer azidation reaction was required to convert both amines to azides. 1H NMR (500 MHz, CDCl3) δ: 7.55 (d, J = 8.5 Hz, 4H), 7.10 (d, J = 8.5 Hz, 4H).
Polymer synthesis
The block copolymer was formed by anionic ring opening polymerization using the PS–OH macroinitiator. First, 50 g of the dry PS–OH was placed in a vacuum flask. To this flask was distilled, under vacuum, 400 mL THF that had been dried over sodium/benzophenone. The PS–OH was dissolved in the THF by stirring. The flask was then removed from the vacuum line using a back flow of dry Ar. Under a flow of Ar, 28.2 mL of a 1.17 molar solution of freshly prepared potassium naphthalide was added by syringe. The flask was sealed, placed back on the vacuum line, and after an hour of stirring, degassed and placed under vacuum. To this was then added 100 g of ethylene oxide (EO) by vacuum distillation. The EO had been dried over calcium hydride for several days, distilled to a flask containing a sodium mirror, distilled to a graduated cylinder, then distilled to the reaction flask at dry ice/isopropanol temperature. The reaction mixture was then sealed, removed from the line, and reacted overnight at room temperature. The reaction was then quenched with methanol, precipitated, and dried. Proton NMR was used to determine the molecular weight and composition.
Before further synthesis, 3 g of the starting copolymer was dissolved in 80 mL toluene, refluxed for 2 h and all solvent was removed in vacuo to remove water as a toluene azeotrope. An alkyne-labeled PS–PEO for subsequent particle surface functionalization was synthesized by treating the PS–PEO (3 g) in an ice bath with sodium hydride (120 mg of 60% dispersion in mineral oil, 3 mmol, ∼6.5 eq.) in 100 mL dry THF for 15 min.25 Subsequently after warming to RT, propargyl bromide (0.28 mL of 80% solution in toluene, 2.83 mmol, ∼6.2 eq.) was added dropwise and the reaction continued at RT for 18 h. The reaction was quenched with water and most of the solvent was removed under reduced pressure. The residue was dissolved in dichloromethane (100 mL) and washed with 100 mL water. After drying the organic layer with Na2SO4, the dichloromethane solution was concentrated to ∼10 mL and dropwise precipitated in 500 mL of ether. The precipitate (2.45 g, ∼81%) was recovered by centrifugation. The appearance of a new doublet in the proton NMR spectrum (δ = 4.21, J = 2.3 Hz) confirmed the addition of the alkyne moiety to the polymer and allowed for an estimate of its molecular weight (Fig. S1†). GPC analysis of this copolymer indicated that it had a molecular weight Mw of 6400, and was highly monodisperse (Mw/Mn = 1.06, Fig. S2†). This alkyne-derivatized polymer was used in all nanoparticle studies described herein.
Assembly of nanoparticles
Alkyne-decorated nanoparticles with either 1 or 2 encapsulated in the core were assembled using Flash NanoPrecipitation (FNP) essentially as described previously.11 In a typical experiment, a THF stream (12 mL min−1) containing PS-b-PEO–alkyne (10 mg mL−1, 1.54 mM, PS mol. wt ≈ 1500, PEO mol. wt ≈ 5000) copolymer, PS–OH (10 mg mL−1, 6.7 mM, mol. wt ≈ 1500) homopolymer, and crosslinker 1 (8.9 mM) or 2 (4.45 mM) was rapidly mixed with three water streams (40 mL min−1 each) in an impinging jet mixer, generating NPs. Following self-assembly of the particles, THF was immediately removed by dialysis against ultrapure water using Spectra/Por dialysis tubing with a molecular weight cutoff of 6000–8000. The dialysis water was changed four times over the course of 24 hours.Mock polymerase chain reaction (PCR) conditions
To test the thermal stability of NPs under typical PCR conditions, particles (90 µL) were mixed with 10 µL 10× Taq buffer to create a 1× working buffer (50 mM KCl, 10 mM Tris–HCl, 1.5 mM MgCl2 and 0.1% Triton X-100). The particles were placed in a Peltier-controlled thermal cycler (Biorad DNA Engine) and subjected to a thermal cycling program composed of 45 cycles of 95 °C for 30 s, 1 °C s−1 ramp to 52 °C, 52 °C for 30 s, 1 °C s−1 ramp to 72 °C, and 72 °C for 75 s, which is a standard protocol used in PCR for DNA amplification. A heated lid was used to minimize evaporation during thermal cycling.PS film crosslinking and 19F NMR analysis
Commercially available polystyrene (Polysciences, reported molecular weight range 800–5000) was used to cast films. GPC analysis of this PS sample revealed two roughly equivalent peaks (Fig. S3†), one with Mw of 1900 and PDI of 1.5 and one with Mw of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and PDI of 2.0. Thus on a number basis, the PS sample contains primarily short PS chains of similar length to the PS chains in the nanoparticle studies described above. However, this commercial sample also contains some contaminating high molecular weight PS as well. To cast PS thin films, a mixture of the PS sample (100 mg) and crosslinker (10 mg, 62.5 µmol for 1, and 42.3 µmol for 2) in toluene (0.5 mL) was spincoated onto a thin glass cover slip treated with trimethoxy(octyl)silane. The center of the slides was exposed to the UV-A lamp (wavelength ≈ 365 nm) for 15 min. The resulting films were dipped in toluene to test for solvent resistance. For NMR analysis, a larger sample was required, so 1 mL of PS/2 mixture (200 mg PS and 8.3 mg (35 µmol) 2) was cast from THF in a 5 cm diameter untreated glass Petri dish. After evaporation of the THF using a nitrogen stream, the film was exposed to UV light for 15 minutes to crosslink the PS. The film was subsequently taken up in CDCl3 and treated with 140 µmol trifluoroacetic anhydride (TFAA) as an acetylating agent for 8 h at RT. As a control, PS with no added crosslinker was subjected to the same irradiation and acetylation. Solvent and residual TFAA were subsequently removed in vacuo. The sample was redissolved in CDCl3 and dried three more times to remove all volatile compounds. Finally, the crosslinked, acetylated PS was dissolved in CDCl3 and its 19F NMR spectrum was acquired using a 300 MHz Varian spectrometer. Spectra were referenced to CFCl3 at 0 ppm. The model compounds diphenylamine 3, N-methylaniline 4, and aniline 5 (70 µmol) were each acetylated with TFAA (140 µmol). Cycles of evaporationin vacuo and resuspension in CDCl3 were carried out to remove excess TFAA. These samples were also analyzed using 19F NMR.
000 and PDI of 2.0. Thus on a number basis, the PS sample contains primarily short PS chains of similar length to the PS chains in the nanoparticle studies described above. However, this commercial sample also contains some contaminating high molecular weight PS as well. To cast PS thin films, a mixture of the PS sample (100 mg) and crosslinker (10 mg, 62.5 µmol for 1, and 42.3 µmol for 2) in toluene (0.5 mL) was spincoated onto a thin glass cover slip treated with trimethoxy(octyl)silane. The center of the slides was exposed to the UV-A lamp (wavelength ≈ 365 nm) for 15 min. The resulting films were dipped in toluene to test for solvent resistance. For NMR analysis, a larger sample was required, so 1 mL of PS/2 mixture (200 mg PS and 8.3 mg (35 µmol) 2) was cast from THF in a 5 cm diameter untreated glass Petri dish. After evaporation of the THF using a nitrogen stream, the film was exposed to UV light for 15 minutes to crosslink the PS. The film was subsequently taken up in CDCl3 and treated with 140 µmol trifluoroacetic anhydride (TFAA) as an acetylating agent for 8 h at RT. As a control, PS with no added crosslinker was subjected to the same irradiation and acetylation. Solvent and residual TFAA were subsequently removed in vacuo. The sample was redissolved in CDCl3 and dried three more times to remove all volatile compounds. Finally, the crosslinked, acetylated PS was dissolved in CDCl3 and its 19F NMR spectrum was acquired using a 300 MHz Varian spectrometer. Spectra were referenced to CFCl3 at 0 ppm. The model compounds diphenylamine 3, N-methylaniline 4, and aniline 5 (70 µmol) were each acetylated with TFAA (140 µmol). Cycles of evaporationin vacuo and resuspension in CDCl3 were carried out to remove excess TFAA. These samples were also analyzed using 19F NMR.
Results and discussion
In addition to NPs encapsulating 1 or 2, several different NP control samples were also generated. First, stable particles were formed when the crosslinker was omitted from the THF stream. Nanoparticles were also stably formed when benzidine (4.45 mM in THF stream), the diamine precursor to 2, serving as a “dummy” crosslinker, was encapsulated in the core. Dynamic light scattering (DLS) analysis indicated that regardless of whether a crosslinker was present, all nanoparticles formed were similar in size, with an average diameter around 120 nm (Fig. 3, filled symbols).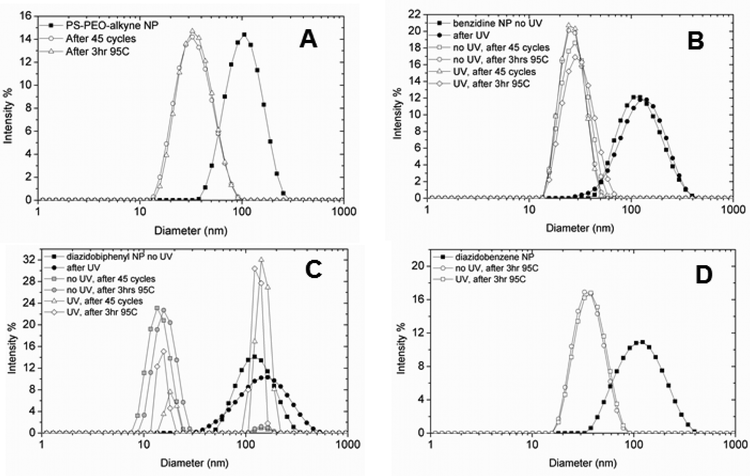 | ||
| Fig. 3 Dynamic light scattering (DLS) analysis of NPs. (A) DLS of NPs containing no encapsulated crosslinker. Solid symbols represent the particle size distribution immediately after Flash NanoPrecipitation and dialysis while open symbols represent particle size after thermal treatment. Following mock PCR conditions or heating at 95 °C, the particles shrink in size due to core mobility under thermal stress. (B) DLS of NPs with dummy crosslinker benzidine encapsulated in the core. Particles shrink in size under thermal stress regardless of whether the particles have been irradiated. (C) DLS of NPs with encapsulated 2. The non-irradiated control particles (gray symbols) shrink in size under mock PCR or constant heating conditions, but the majority of irradiated NPs (open symbols) remain the same size under these conditions. (D) Disassembly of NPs with core-encapsulated 1 upon thermal challenge at 95 °C for 3 h. Both the irradiated sample (triangles) and non-irradiated control sample (circles) shrink in size from ∼120 nm to 30 nm in response to thermal challenge. | ||
We next turned our attention to photoinitiating the crosslinkers in the core of the NPs. Following formation of the NPs, an aqueous suspension of the particles was placed in a Petri dish and irradiated with a UV-A lamp (λ ≈ 365 nm) for 15 min. While suspensions of the control nanoparticles containing either no crosslinker or the dummy crosslinker benzidine retained their whitish hue after irradiation, particles with 2 encapsulated became bright yellow after irradiation (Fig. 4). There was also a less drastic color change noted after UV irradiation for particles with encapsulated 1 (data not shown).
 | ||
| Fig. 4 Color change upon irradiation of NPs with 2 encapsulated in the core. From left, I: PS-b-PEO NPs with no crosslinker in the core, II: NPs with dummy crosslinker benzidine in the core, III: UV-irradiated NPs with benzidine in the core, IV: NPs with 2 in the core, V: UV-irradiated NPs with 2 in the core. | ||
To assess the performance of crosslinkers 1 and 2 in endowing thermal stability to NPs, we subjected our UV-irradiated PS-b-PEO–alkyne NPs containing crosslinker, non-irradiated controls, and NPs without any crosslinker to a mock polymerase chain reaction (PCR). A standard thermal cycling program for PCR was carried out as described in the Experimental section. As a more stringent measure of thermal stability, we also incubated the NP samples at 95 °C in a thermal cycler for 3 hours. As shown in Fig. 3, NPs without encapsulated crosslinkers disassembled and reassembled into smaller polymeric micelles with sizes on the order of 30 nm after rapid thermal cycling or a 3 h 95 °C incubation (Fig. 3A). This thermally induced micellization of PS-b-PEO chains has been previously reported by Wilhelm et al.26 NPs encapsulating benzidine also disassembled during the thermal cycling of the mock PCR, regardless of whether the particles were irradiated (Fig. 3B). In contrast, the majority of irradiated nanoparticles with 2 in the core retained their initial size following either the mock PCR conditions or heating for 3 h at 95 °C (Fig. 3C, open triangles and diamonds). Irradiation of the particles was critical in establishing their thermal stability; non-irradiated particles with 2 in the core (Fig. 3C, grey squares and circles), shrank in size upon thermal cycling, with only a small population staying intact. UV-irradiated PS-b-PEO–alkyne NPs with 1 encapsulated in their cores were not as stable under thermal challenge as NPs with 2 encapsulated (Fig. 3D).
In limit of very long polymer chains, one crosslinker per two chains, the stoichiometry used in our experiments above, is sufficient to generate an infinite network. However, in our case the PS chains are quite short, with an average chain length of ∼15. The reaction of the diazide 2 with the 15-mer polystyrene can be analyzed using Flory's approach to crosslinking of polymer chains.27 For a crosslinked polymer chain of N0 total monomer units with ν/2 crosslinkages, the crosslinking density ρ is given by the following:
| ρ = ν/N0 |
| ε = ρ(y − 1) |
We were also interested in whether 1 and 2 could function in environment other than a confined NP core, so we tested their ability to crosslink thin films of polystyrene. Additionally, films composed of PS only are a more tractable system for the NMR analysis we describe below. Upon irradiation, the film with 1 turned light brown and the film with 2 turned yellow, consistent with the color changes we observed for the particles. The solvent resistance of the films was assessed by dipping the cover slip into toluene for 15 s. While this treatment washed off the majority of the uncrosslinked PS, crosslinker 2 provided significant resistance to toluene (Fig. 5). Although a color change indicated some reaction of crosslinker 1, the irradiated PS + 1 film did not exhibit the same solvent resistance as the PS + 2 film.
 | ||
| Fig. 5 Solvent resistance of polystyrene (PS) films crosslinked by 1 and 2. Left: the center region of the film was exposed to a UV lamp for 15 min, resulting in a color change in the film. Right: the films were dipped in toluene, an excellent solvent for PS, for 15 s and imaged again. Crosslinker 2 provides some resistance to this solvent treatment. | ||
Since the nitrene moiety which arises from photolysis of aryl azides is known to insert into aliphatic C–H bonds but not aromatic C–H bonds,23 we expected that 2 would function as a crosslinker by bridging carbon atoms within the PS backbone, generating a secondary amine (Fig. 6). Without any crosslinking, the aryl azide functionality would degrade into primary amine groups upon photolysis. The difference in chemical shift between the primary amine and secondary amine acetylation products was probed by performing acetylation with trifluoroacetic anhydride on model compounds, including diphenylamine, N-methylaniline and aniline (Fig. 6). Diphenylamine (3) is a model compound corresponding to the insertion of 2 into the phenyl ring of PS thus generating a diaryl amine while N-methylaniline (4) is a model for the insertion of 2 into the PS backbone (Fig. 6) and aniline (5) is a model for degradation of 2 to a primary amine. Such a structure could arise in the PS films in a failed crosslinking event. For instance, if one nitrene inserted into PS but the other nitrene was quenched by solvent before it could insert into PS, the resulting structure would contain one secondary amine and one aniline-like primary amine.
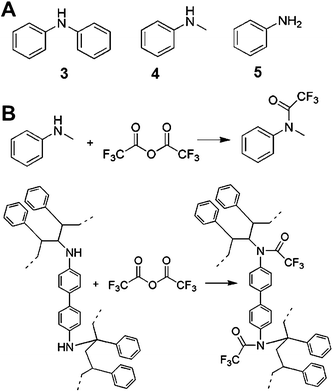 | ||
| Fig. 6 Examination of the chemical structure of PS crosslinked by 2. (A) Model compounds corresponding to insertion of 2 into the PS phenyl ring (3) or the PS backbone (4). Compound 5 is a model compound for ε = ρ(y − 1) that was quenched before insertion into PS. (B) top: acetylation of the model compound 4 by trifluoroacetic anhydride (TFAA), bottom: TFAA acetylation of PS chains crosslinked by 2. | ||
Upon acetylation of the model compounds with TFAA, the difference in the 19F chemical shift between primary and secondary amines is readily apparent with the diphenylamine and N-methylaniline peaks showing up as singlets at −67.28 ppm and −67.47 ppm, respectively, and the aniline peak much further upfield at −76.15 ppm (Fig. 7). These values agree with previously reported chemical shifts of the trifluoroacetyl group attached to primary and secondary amines.28 With these values in hand, we turned our attention to the nature of the PS crosslinking with 2. PS films photolyzed in the presence of 2 and treated subsequently with TFAA exhibit a peak at −68.10 ppm demonstrating the presence of secondary amines (Fig. 7). Since 19F chemical shifts have a strong dependence on the chemical environment,29 we cannot unequivocally assign this peak to an N-methylaniline-like structure, though the reported reactivity of aryl nitrenes23 suggests that the nitrene formed by 2 should insert into the methylene or methine groups of the PS backbone but not the aromatic rings of PS. No such peak appears in an NMR spectrum of uncrosslinked PS treated with TFAA (Fig. 7), though three intense upfield peaks appear in this sample. These peaks are likely from the incomplete removal of TFAA or its hydrolysis product trifluoroacetic acid (TFA) from the PS samples. Several additional upfield resonances are observed in the 19F spectrum of the crosslinked PS relative to the uncrosslinked material including small peaks at −75.17, −75.26, and −75.98 ppm. We hypothesize that these peaks may arise from acetylation of primary amines resulting from the failed crosslinks discussed above. Collectively, these NMR experiments demonstrate that 2 forms a network in PS by generating crosslinks composed of secondary amines.
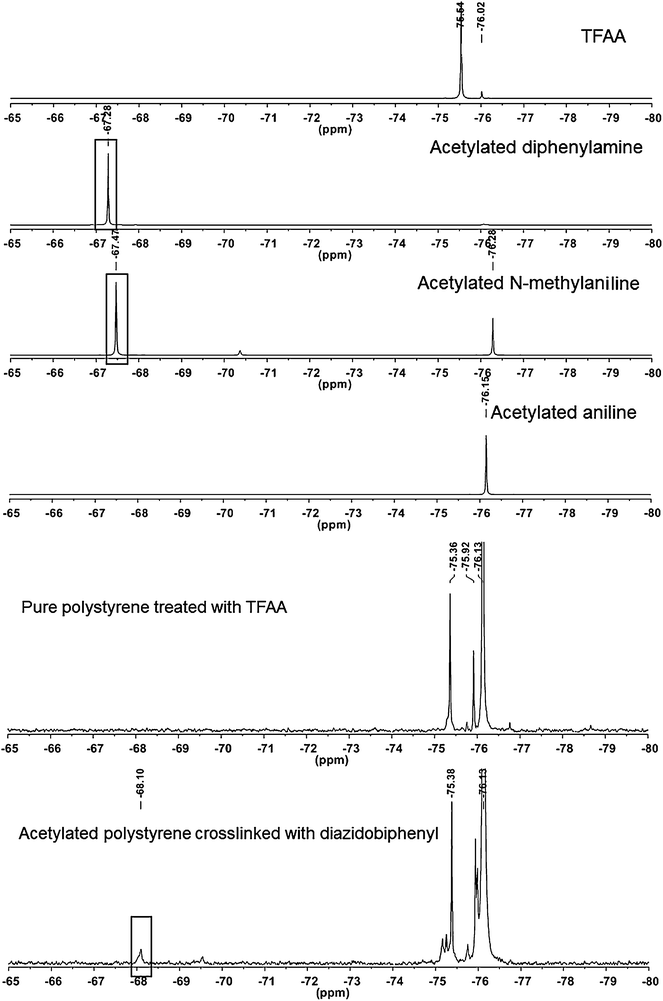 | ||
| Fig. 7 19F NMR of TFAA acetylated model compounds, pure PS, and PS crosslinked by 2. Peaks corresponding to the acetylation of secondary amines are highlighted by boxes. | ||
Conclusion
In this paper we have demonstrated that the aryl diazide compound 4,4′-diazidobiphenyl 2 can serve as an efficient photoinitiated crosslinker when it is encapsulated in the core of PS-b-PEO block copolymer NPs. Upon UV irradiation, suspensions of these NPs undergo a drastic color change from whitish to bright yellow. Furthermore, these NPs exhibit much improved thermal stability as compared to their uncrosslinked counterparts. Compound 2 can also crosslink spun-cast polystyrene films, endowing these films with improved solvent resistance. While 2 serves as a good crosslinker, the smaller aromatic diazide 1 does not function well in this regard. One likely reason as to why 2 performs better is that the two azide moieties are spaced farther apart in 2 than in 1 and are thus more electronically distinct; in other words, the two reactive nitrenes can form and react independently. Alternatively, the greater length between the azides could allow the crosslinker to more readily span the distance between two PS chains either within the core of a nanoparticle or in a thin film. In addition to being a novel compound for the stabilization of nanoparticles, compound 2 and other related polycyclic aromatic diazides should be able to serve as photocrosslinkers in nano-confined regimes for a wide variety of hydrophobic polymers.Acknowledgements
This work was supported by a seed grant from the NSFMRSEC-funded Princeton Center for Complex Materials and by National Science Foundation Grant NIRT CBET-0506966. We thank Abigail Doyle and Rodney Priestley for helpful discussions.References
- F. X. Gu, R. Karnik, A. Z. Wang, F. Alexis, E. Levy-Nissenbaum, S. Hong, R. S. Langer and O. C. Farokhzad, Nano Today, 2007, 2, 14–21 CrossRef.
- M. E. Gindy and R. K. Prud'homme, Expert Opin. Drug Delivery, 2009, 6, 865–878 Search PubMed.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2008, 83, 761–769 CrossRef CAS.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2008, 19, 1423–1429 CrossRef CAS.
- M. Akbulut, P. Ginart, M. E. Gindy, C. Theriault, K. H. Chin, W. Soboyejo and R. K. Prud'homme, Adv. Funct. Mater., 2009, 19, 718–725 CrossRef CAS.
- M. E. Gindy, S. Ji, T. R. Hoye, A. Z. Panagiotopoulos and R. K. Prud'homme, Biomacromolecules, 2008, 9, 2705–2711 CrossRef CAS.
- T. Musacchio, V. Laquintana, A. Latrofa, G. Trapani and V. P. Torchilin, Mol. Pharmaceutics, 2009, 6, 468–479 CrossRef CAS.
- B. K. Johnson and R. K. Prud'homme, Aust. J. Chem., 2003, 56, 1021–1024 CrossRef CAS.
- B. K. Johnson and R. K. Prud'homme, Phys. Rev. Lett., 2003, 91, 118302-1–118302-4.
- Y. Liu, K. Kathan, W. Saad and R. K. Prud'homme, Phys. Rev. Lett., 2007, 98, 036102-1–036102-4.
- M. E. Gindy, A. Z. Panagiotopoulos and R. K. Prud'homme, Langmuir, 2008, 24, 83–90 CrossRef CAS.
- S. Brenner, M. Johnson, J. Bridgham, G. Golda, D. H. Lloyd, D. Johnson, S. J. Luo, S. McCurdy, M. Foy, M. Ewan, R. Roth, D. George, S. Eletr, G. Albrecht, E. Vermaas, S. R. Williams, K. Moon, T. Burcham, M. Pallas, R. B. DuBridge, J. Kirchner, K. Fearon, J. Mao and K. Corcoran, Nat. Biotechnol., 2000, 18, 630–634 CrossRef CAS.
- B. R. Graveley, Nature, 2008, 453, 1197–1198 CrossRef CAS.
- N. Cloonan, A. R. R. Forrest, G. Kolle, B. B. A. Gardiner, G. J. Faulkner, M. K. Brown, D. F. Taylor, A. L. Steptoe, S. Wani, G. Bethel, A. J. Robertson, A. C. Perkins, S. J. Bruce, C. C. Lee, S. S. Ranade, H. E. Peckham, J. M. Manning, K. J. McKernan and S. M. Grimmond, Nat. Methods, 2008, 5, 613–619 CrossRef CAS.
- S. C. Schuster, Nat. Methods, 2008, 5, 16–18 CrossRef CAS.
- N. J. Clecak, R. J. Cox and W. M. Moreau, Polym. Eng. Sci., 1974, 14, 491–493 CrossRef CAS.
- A. Yen, H. I. Smith, M. L. Schattenburg and G. N. Taylor, J. Electrochem. Soc., 1992, 139, 616–619 CrossRef CAS.
- D. Roy, P. K. Basu, P. Raghunathan and S. V. Eswaran, Magn. Reson. Chem., 2003, 41, 671–678 CrossRef CAS.
- S. X. Cai, D. J. Glenn, M. Kanskar, M. N. Wybourne and J. F. W. Keana, Chem. Mater., 1994, 6, 1822–1829 CrossRef CAS.
- B. T. Storey, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 265–282 CrossRef CAS.
- H. M. Tsay and R. Ullman, Macromolecules, 1988, 21, 2963–2972 CrossRef CAS.
- W. R. Bussing and N. A. Peppas, Polymer, 1983, 24, 209–216 CrossRef CAS.
- G. B. Schuster and M. S. Platz, Adv. Photochem., 1992, 17, 69–143 CAS.
- J. Miyake and Y. Chujo, Macromolecules, 2008, 41, 5671–5673 CrossRef CAS.
- S. Schoffelen, M. H. L. Lambermon, M. B. van Eldijk and J. C. M. van Hest, Bioconjugate Chem., 2008, 19, 1127–1131 CrossRef CAS.
- M. Wilhelm, C. L. Zhao, Y. C. Wang, R. L. Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033–1040 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, London, 1953 Search PubMed.
- J. M. Skutnik, R. A. Assink and M. Celina, Polymer, 2004, 45, 7463–7469 CrossRef CAS.
- M. A. Danielson and J. J. Falke, Annu. Rev. Biophys. Biomol. Struct., 1996, 25, 163–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GPC and NMR analysis of PS-b-PEO-alkyne block copolymer and GPC analysis of commercial PS sample. See DOI: 10.1039/c0py00350f |
| This journal is © The Royal Society of Chemistry 2011 |