Computational study of esterification between succinic acid and ethylene glycol in the absence of foreign catalyst and solvent†
Tao
Yu
*,
Hai-Bo
Chang
,
Wei-Peng
Lai
and
Xiao-Fang
Chen
Computer-Aided Energetic Materials Design Group, Xi'an Modern Chemistry Research Institute, Xi'an, P. R. China. E-mail: fischer@wo.com.cn; fischer@wo.com.cn; Tel: (+86) 029-88294020
First published on 12th January 2011
Abstract
The reaction mechanisms, thermodynamic and kinetic properties of polyesterification in the absence of foreign catalyst and solvent between succinic acid and ethylene glycol were investigated by the density functional theory. The polyesterification proceeds by the concerted and stepwise mechanisms, involved in the self catalysis and non-self catalysis respectively. The self catalysts include acid, alcohol and water. The geometries of reactants, transition states, and intermediates were optimized at the B3LYP/6-31G** level. The vibrational analysis was carried out to confirm the transition state structures, and the intrinsic reaction coordinate (IRC) method was used to explore the energy path. The statistical thermodynamic method and Eyring transition state theory with Wigner correction were used to investigate the thermodynamic and kinetic characters of four reaction channels. The first step with self acid catalyst, the optimum channel, was taken for example. The reaction rate constant increases as the temperature rises and there is little influence of pressure in theoretical and computational conditions. The transition from singlet state to triplet state is probably forbidden. The same degree of polymerization (DP) of self acid catalysts has similar catalysis in the different reaction systems. The catalysis of different self acid catalysts may be similar for the same reaction system which doesn't include self catalyst and dimerization is probably the rate-limiting procedure.
Introduction
Esterification between dibasic acid and glycol is an important class of chemical reaction in polymer chemistry. The oligoesters are univerally synthesized without outside catalyst and solvent, because they are not easily separated from the products and quality control is not conveniently realized in complex reaction systems.Works1–3 by Flory have been widely recognized. According to Flory, self catalysis polyesterification in the absence of a foreign acid, owing to a second molecule of acid undergoing irreversible esterification as catalyst, is a third-order reaction. Tang and Yao4 indicated the reaction is 2.5-order, acid and alcohol make co-catalysis. Lin and Hsieh5 deduced second-order on alcohols and first-order on acid. Atkins' Physical Chemistry6 gives an example of first-order on acid and first-order on alcohol too. Salmi et al.7 surveyed kinetic model for the increase of reaction order during polyesterification. Lehmus et al.8 obtained the complex mechanisms.
The classical mechanism of Fischer–Speier esterification9,10 in the presence of a foreign acid catalyst has five steps: proton transfer from acid catalyst to carbonyl oxygen increases the electrophilicity of the carbonyl carbon; the carbonyl carbon is then attacked by the nucleophilic oxygen atom of the alcohol; proton transfer from the oxonium ion to a second molecule of the alcohol gives an activated complex; protonation of one of the hydroxyl groups of the activated complex gives a new oxonium ion; loss of water from this oxonium ion and subsequent deprotonation gives the ester.11 The mechanism can be also referred to for esterification in the absence of a foreign acid catalyst.
To the best of our knowledge, no computational study on polyesterification in the absence of foreign catalyst and solvent between succinic acid and ethylene glycol was reported. In this paper, two possible mechanisms, that is, the concerted and stepwise mechanisms, have been put forward. The self catalysis has been investigated in detail, compared with the non-self catalysis.
Computational details
Research on chemical reaction mechanisms is usually realized by kinetics and transition states.12,13 The chemical formula of the reactants (R), transition states (TS), intermediates (IM), and products (P) in four reaction channels shown in Fig. 1 were optimized using the Gaussian 09 software package14 at the B3LYP/6-31G**15–17 level. The method and basis set have been well used in transition states of condensation reactions.18,19 The structures and imaginary frequencies of transition states were confirmed by the vibration analysis calculation and the intrinsic reaction coordinate (IRC) calculation at the same level. The statistical thermodynamic method and Eyring transition state theory6,20–22 with Wigner correction23 (eqn (1) and (2)) were used to determine the thermodynamic functions and rate constants of all mechanisms.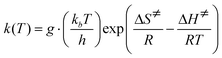 | (1) |
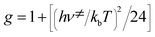 | (2) |
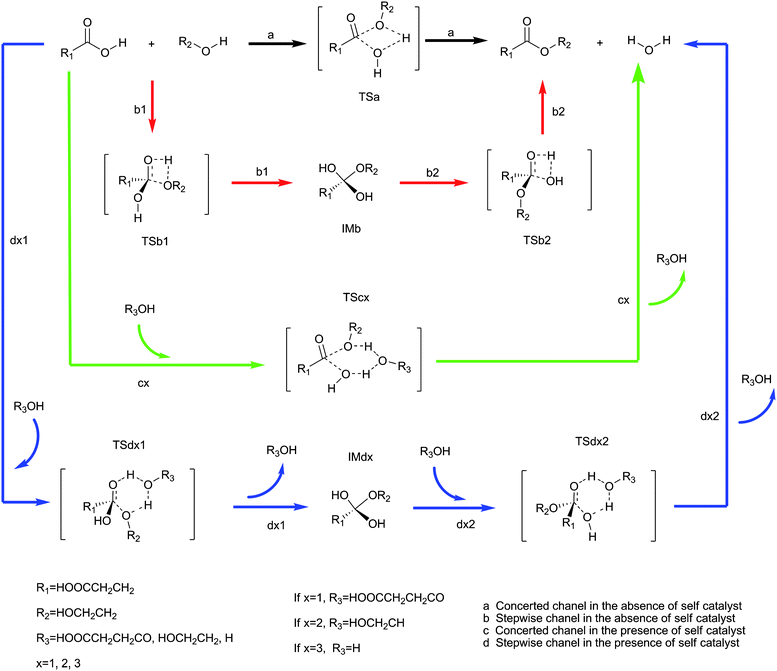 | ||
| Fig. 1 Possible mechanisms for esterification between succinic acid and ethylene glycol. The IM and TS represent intermediates and transition states respectively. a, b, c and d represent the channels. 1 and 2 represent the first and second step respectively. The first step is an addition reaction and the second step is a dehydration reaction. x represents catalysts, when x = 1, it is acid, when x = 2, it is alcohol, when x = 3, it is water. Channel a: black. Channel b: red. Channel c: green. Channel d: blue. | ||
Results and discussion
Activation energy barriers
According to the mechanisms made by Fischer–Speier9 and Patel,10 the transition states without self catalyst were designed to C–O–H–O tetra-atomic ring geometry and the transition states with self catalyst were designed to C–O–H–O–H–O hex-atomic ring geometry, see Fig. 1. The key bond lengths of transition states are presented in the ESI.† The activation energy barriers for all mechanisms, in the absence of foreign catalyst and solvent, are shown in Fig. 2 and potential energy profiles of reverse channels are presented in the ESI.† The results are originally from IRC at B3LYP/6-31G** level. There is rotational isomerism in the reactants, intermediates, and products. In channel a, see Fig. 1, the carbonyl carbon is attacked by the oxygen atom of the alcohol and the water is lost from hydroxyl group of acid and hydrogen atom of alcohol. In channel b1, the proton transfer from alcohol to carbonyl oxygen and the carbonyl carbon is attacked by oxygen atom of the alcohol. In channel b2, the water is lost from the condensation of two hydroxyl groups. The succinic acid and ethylene glycol reacting directly without self catalyst, channel a and b, shows the higher energy barriers. The concerted mechanism, channel a, is 131.27 kJ/mol. The first step, channel b1, is 136.29 kJ/mol and the second step, channel b2, is 137.44 kJ/mol in stepwise mechanism. All of the activation energy barriers are above 130 kJ/mol. In channel c, see Fig. 1, the carbonyl carbon is attacked by the oxygen atom of the alcohol and the self catalyst, acid, alcohol or water, links to the acid and alcohol as “bridge”. The water is lost from the hydroxyl group of the acid and a hydrogen atom of the self catalyst and a hydrogen atom is compensated to the self catalyst by the alcohol. In channel d1, the proton transfers from self catalyst to carbonyl oxygen and the carbonyl carbon is attacked by the oxygen atom of the alcohol. The hydrogen atom is also compensated to the self catalyst by the alcohol on the “bridge”. In channel d2, the water is lost on the “bridge”. The highest energy barrier, 138.99 kJ/mol, is in the concerted mechanism with self acid catalyst, channel c1. But the energy barriers are distinctly lower in the stepwise mechanism, the first step, channel d11, is 79.27 kJ/mol and the second step, channel d12, is 60.22 kJ/mol. There are moderate activation energy barriers in the self alcohol catalyst mechanism. The concerted mechanism, channel c2, is 95.68 kJ/mol, the first step, channel d21, is 92.62 kJ/mol and the second step, channel d22, is 73.42 kJ/mol in the stepwise mechanism. The energy barriers with the water catalyst are 111.40 kJ/mol in the concerted mechanism, channel c3, the first step, channel d31, is 96.92 kJ/mol and the second step, channel d32, is 78.17 kJ/mol in the stepwise mechanism. The activation energy barrier was reported2 to be about 100 kJ/mol. The energy barriers of channel a, b and c1 are higher than the reported value by 30 kJ/mol. The energy barriers are lowered effectively except channel c1 by the “bridge”.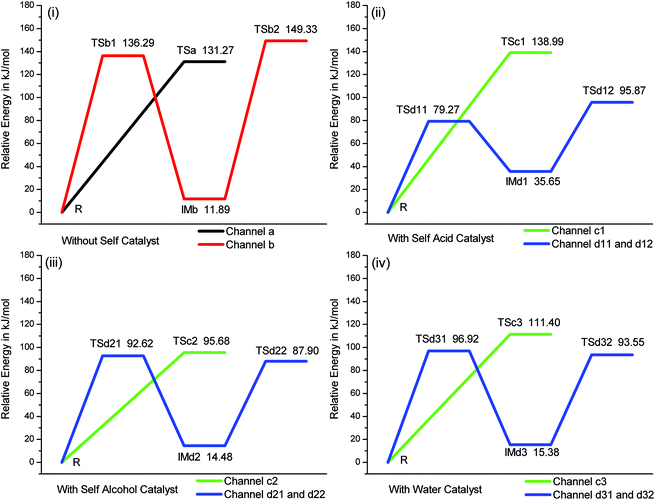 | ||
| Fig. 2 The potential energy profiles of four channels. (i) In the absence of self catalyst. (ii) In the presence of acid self catalyst. (iii) In the presence of alcohol self catalyst. (iv) In the presence of water catalyst. Channel a: black. Channel b: red. Channel c: green. Channel d: blue. | ||
Thermodynamics and kinetics
The experimental research2 on polyesterification between succinic acid and ethylene glycol without solvent was studied at 123 °C. The reaction rate constant for first-order, deduced by us based on ref. 2, is 2.22 × 10−4 s−1 when the fractional conversion equals 0.5. The thermodynamics and kinetics performance for all mechanisms is presented in Table 1. The mechanism with self acid catalyst has the distinctly lower imaginary frequency and less Wigner emendation factor. The reaction rate constants without self catalyst are the lowest in both concerted and stepwise reactions. The rate constants of concerted reactions are generally larger than those of stepwise reactions. The first step, addition, is clearly rate-limiting for the reaction between succinic acid and ethylene glycol. The second step, dehydration, has fast reaction rate for self acid, self alcohol and water catalyst. The optimum channel is stepwise mechanism with self acid catalyst, channel d1. The reaction rate constant for the first step, 2.91 × 10−4s−1, is closest to the experimental data. But the concerted mechanism with self acid catalyst has the higher activation energy barrier and less reaction rate constant than with self alcohol and water. The materials of acid and alcohol are not equal mole, always the alcohol is excess, for synthesis of polyester polyol. In the later period of reaction, hydroxyl groups are much more than carboxyl groups, and the water is released from reaction system. The self alcohol catalyst may play the leading role, but the reaction rate should slow down.| Reactions | ΔH≠ | ΔS≠ | ν ≠ | g | k(T) |
|---|---|---|---|---|---|
| a The unit of ΔH≠ is kJ mol−1, the unit of ΔS≠ is kJ mol−1 K−1, the unit of ν≠ is cm−1, the unit of k(T) is s−1. | |||||
| R → TSa | 153.34 | −65.24 | 1149.48i | 1.73 | 3.36 × 10−11 |
| R → TSb1 | 158.80 | −51.57 | 1537.24i | 2.30 | 4.41 × 10−11 |
| IMb → TSb2 | 126.67 | −2.54 | 1605.03i | 2.42 | 2.91 × 10−4 |
| R → TSc1 | 154.06 | −47.06 | 434.57i | 1.10 | 1.54 × 10−10 |
| R → TSd11 | 92.30 | −85.18 | 312.18i | 1.05 | 2.08 × 10−4 |
| IMd1 → TSd12 | 52.08 | −19.04 | 587.69i | 1.19 | 1.35 × 105 |
| R → TSc2 | 129.75 | −69.84 | 1138.79i | 1.71 | 2.47 × 10−8 |
| R → TSd21 | 103.68 | −92.69 | 1335.81i | 1.98 | 5.02 × 10−6 |
| IMd2 → TSd22 | 58.21 | −34.16 | 1339.44i | 1.99 | 5.68 × 103 |
| R → TSc3 | 127.22 | −79.82 | 1216.49i | 1.81 | 1.70 × 10−8 |
| R → TSd31 | 103.32 | −89.21 | 1360.39i | 2.02 | 8.66 × 10−6 |
| IMd3 → TSd32 | 59.25 | −24.42 | 1383.58i | 2.05 | 1.38 × 104 |
Temperature, pressure and triplet state
The first step with self acid catalyst was taken for example to discuss the influences of temperature and pressure on reaction rate constants. The imaginary frequency of transition state has no change, the activation enthalpy and activation entropy decrease when the temperature increases. The relationship between mole activation Gibbs free energy and temperature is linear, see Fig. 3. The first derivation of eqn (1) and (2) for temperature can be expressed as: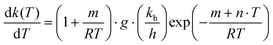 | (3) |
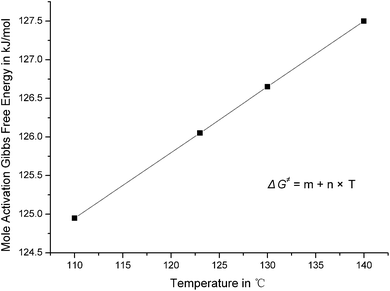 | ||
| Fig. 3 The linear relationship between mole activation Gibbs free energy and temperature in the first step with self acid catalyst. | ||
The reaction for polyester polyol generally goes on negative pressure condition at the later period.24–26 The influence of pressure on reaction rate constant is caused by removal of water leading to the break of reaction equilibrium. But there is little influence on rate constant in quantum chemistry research, because it is single molecular system for reactant and transition state. The computational results surveyed the viewpoint. There is no change on activation enthalpy and imaginary frequency of transition state, although the changes are made on absolute values of activation entropy, not on relative values.
The triplet state is very important in chemical reactions.27,28 Our calculations found that the energies of reactant and product with triplet state in the first step of the optimum channel are higher than singlet of transition state by about 3 eV. Furthermore, the transition from singlet state to triplet state is forbidden. So the triplet state is probably not involved in the reaction system.
Degree of polymerization (DP)
DP is an important parameter in polymer science. It is defined as the total number of structural units29 and related to the fractional conversion in polyesterification, kinetics and molecular weight. DP also correlates with physical properties such as melting temperature and mechanical strength.30 The first step with self acid catalyst was also taken as an example to discuss the influences of different DP on activation energy barriers, see Fig. 4. In the same molecular system, the activation energy barrier of 2-1-2 differs from 2-2-1 in about 1 kJ/mol. It is similar to 3-1-2 and 3-2-1. It shows that there is interactive self catalysis in the same molecular system. The activation energy barrier of 2-1-2 and 2-2-1 are approximately equal to 3-1-2 and 3-2-1 respectively. It shows that the same DP of self acid catalysts has similar catalysis in the different reaction systems. The activation energy barrier of 1-1-1 is not more than 1 kJ/mol with 1-1-3. The catalysis of different acids may be similar for the same reaction system, not including self catalyst. All of the above partly indicate the equal activity of functional groups.1 The activation energy barriers of reaction between succinic acid and ethylene glycol are higher than a in high DP system. The dimerization is probably the rate-limiting procedure. After dimerization, the reaction goes into a smooth phase.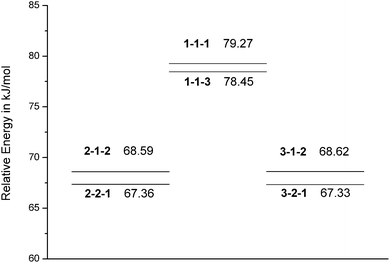 | ||
| Fig. 4 The potential energy profile of different degree of polymerization (DP) in the first step with self acid catalyst. The first number represents the DP of alcohol; the second number represents the DP of acid; the third number represents the DP of self acid catalyst. | ||
Conclusions
The optimum channel of esterification between succinic acid and ethylene glycol is stepwise mechanism with self acid catalyst and dimerization is probably the rate-limiting procedure. We suggest that the acid should be excess for synthesis of polyester polyol at the initial period and the alcohol should be added step by step. The reaction rate can be accelerated with self acid catalyst and the terminal groups can be blocked by hydroxyl groups.The reaction rate constant increases as the temperature rises. But the temperature can't be higher than 164 °C, the boiling point of succinic acid, at the initial period. High temperature means that high performance of apparatus, such as oil–water separator—removal of water, reflux condenser—avoiding loss of reactants and nitrogen gas—protecting the reaction system, are required. When the fractional conversion of hydroxyl group gets to about 0.7, the rising temperature step by step is an effective method to accelerate the reaction rate. The influences of pressure on polyesterification are a break in chemical equilibrium and take low molecular weight polyester away, then the hydroxyl value is lowered. But there is little influence in theoretical and computational conditions.
More than several times excess alcohol or acid in the reaction system is generally applied in kinetic research of polyesterification. But the self alcohol and self acid catalysis show different mechanisms in our research. Which leads the reaction is the primary issue.
Notes and references
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press Ithaca, New York, 1953 Search PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1937, 59, 466 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1939, 61, 3340 CrossRef.
- A.-C. Tang and K.-S. Yao, J. Polym. Sci., Chem. Ed., 1959, 35, 219 Search PubMed.
- C. C. Lin and K. H. Hsieh, J. Appl. Polym. Sci., 1977, 21, 2711 CrossRef CAS.
- P. Atkins and J. D. Paula, Atkins' Physical Chemistry, Oxford University Press, London, 8th edn, 2006 Search PubMed.
- T. Salmi, E. Paatero and P. Nyholm, Chem. Eng. Process., 2004, 43, 1487 CrossRef CAS.
- M. O. Lehmus, S. Toppinen, M. K. Seläntaus, N. M. Kopola and A. O. I. Krause, Ind. Eng. Chem. Res., 1999, 38, 4250 CrossRef CAS.
- E. Fischer and A. Speier, Ber. Dtsch. Chem. Ges., 1895, 28, 3252 CrossRef CAS.
- S. Naik, V. Kavala, R. Gopinath and B. K. Patel, ARKIVOC, 2006, i, 119.
- R. C. Larock, Comprehensive Organic Transformations, VCH, New York, 1989 Search PubMed.
- K. J. Laidler and M. C. King, J. Phys. Chem., 1983, 87, 2657 CrossRef CAS.
- J. C. Polanyi, Science, 1987, 236, 680 CrossRef CAS.
- Gaussian 09, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Regna, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab Initio Molecular Obital Theory, Wiley, New York, 1986 Search PubMed.
- P. Lian, W.-P. Lai, B.-Z. Wang, Z.-X. Ge and Y.-Q. Xue, A density functional theory study on diazotization and nitration of 3,5-diamino-1,2,4-triazole, Int. J. Quantum Chem. DOI:10.1002/qua.22517.
- X.-F. Chen, C.-Y. Hou and K.-L. Han, J. Phys. Chem. A, 2010, 114, 1169 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 107 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875 RSC.
- D. J. Winzor and C. M. Jackson, J. Mol. Recognit., 2006, 19, 389 CrossRef CAS.
- E. Wigner, Phys. Rev., 1932, 40, 749 CrossRef CAS.
- C. J. Maurer, US Patent, 4670580, 1987.
- J.-Y. Wang and X.-H. Ni, Int. J. Plant Eng. Manag., 2003, 44, 60 Search PubMed.
- F.-C. Chang, Handbook of Applied Polymers, Wunan Book Co., Ltd., Taipei, 2006 Search PubMed.
- T.-S. Chu, Y. Zhang and K.-L. Han, Int. Rev. Phys. Chem., 2006, 25, 201 CrossRef CAS.
- K.-L. Han and G.-Z. He, J. Photochem. Photobiol., C, 2007, 8, 55 CrossRef CAS.
- IUPAC Compendium of Chemical Terminology, 2nd edn, 1997 Search PubMed.
- P. J. Flory and A. Vrij, J. Am. Chem. Soc., 1963, 85, 3548 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Key bond lengths of transition states and potential energy profiles of reverse channels. See DOI: 10.1039/c0py00381f |
| This journal is © The Royal Society of Chemistry 2011 |