Conjugated alternating copolymers with 4,4′-dimesitylboryl-2,2′-bithiophene as a building block†
Haiyan
Li
and
Frieder
Jäkle
*
Department of Chemistry, Rutgers University-Newark, 73 Warren Street, Newark, NJ 07102, USA. E-mail: fjaekle@rutgers.edu
First published on 24th December 2010
Abstract
The functionalization of polythiophene with dimesitylborane π-acceptor groups has a strong effect on the conjugated polymer backbone, resulting in interesting optical and electronic properties. To investigate the interplay between the electron-deficient organoborane substituents and the conjugated polymer main chain, a series of alternating copolymers was prepared. Several aromatic building blocks that are commonly used in organic semiconductors, i.e.fluorene, carbazole, and triphenylamine, were chosen as comonomers for combination with 4,4′-diboryl-2,2′-bithiophene units. First, the trimethylsilyl-substituted copolymers PFT-Si, PCT-Si, and PTT-Si were prepared via standard Suzuki coupling reaction of 5,5′-diiodo-4,4′-bis(trimethylsilyl)-2,2′-bithiophene and the respective pinacolborane-functionalized aromatic comonomer. Subsequent post-polymerization modification reactions, which involved treatment with BBr3 and then mesitylcopper, led to three borylated copolymers, PFT-B, PCT-B, and PTT-B. The degrees of polymerization (DPn) and the polydispersities are consistent with those of the silylated precursor polymers. The photophysical and electrochemical properties of these new polymers have been studied in detail.
Introduction
Over the past several decades, conjugated polymers have become one of the most rapidly growing fields in material science because of their potential applications, for example, in light emitting devices, field effect transistors, photovoltaics, chemical and biological sensors, etc.1 Relative to their inorganic counterparts, polymeric materials offer several advantages including flexibility, light weight, and solution processability. Especially for thiophene-based materials, the optoelectronic properties can be manipulated easily through a vast array of chemical structure variations and modifications with different functional groups.2 One such modification that can lead to significant changes in the properties is the introduction of metals or more generally inorganic elements.3 Recently, several straightforward routes for the incorporation of tricoordinate organoborane moieties into conjugated polymers have been successfully developed.4 Triorganoborane moieties are of particular interest because they can function as electron acceptors and the empty p orbital on boron facilitates π-delocalization.5–10 The presence of an empty p orbital also typically leads to high Lewis acidity of triorganoboranes, which is advantageous for the detection of anions and toxic chemicals such as fluoride and cyanide species.6,8,11In our pursuit of new conjugated polymeric materials with interesting and unusual properties, we recently prepared the organoborane-functionalized polythiophene PT-B from PT-Sivia a polymer modification strategy (Chart 1).7 Electrochemical data showed that PT-B has a much lower-lying LUMO level than the silylated precursor PT-Si due to the electron deficient nature of the dimesitylboryl substituents. In related work, Yamaguchi and coworkers9 developed dimesitylboryl-substituted aryleneethynylene-based oligomers and polymers, which show intense emission in both solution and the solid state, and Lambert and Reitzenstein10 reported on borylated polycarbazoles, in which electron-rich arylamine groups in the conjugated polymer main chain are combined with electron-deficient dimesitylboryl substituents. To investigate the effect of modifications in the main chain skeleton on the electronic structure and the optical properties of dimesitylboryl-functionalized polythiophenes, we have now prepared a series of copolymers, in which borylated bithiophene units alternate with other organic π building blocks, specifically fluorene, carbazole, and triphenylamine derivatives.
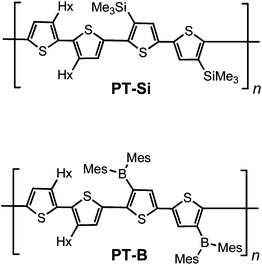 | ||
| Chart 1 | ||
Results and discussion
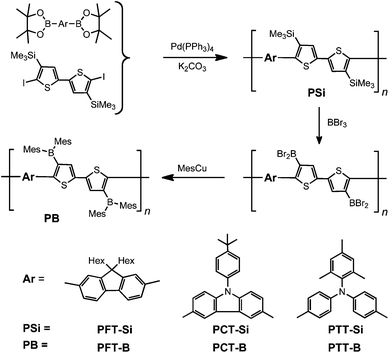
A series of silylated bithiophene-based alternating copolymers were prepared by standard Suzuki coupling as shown in Scheme 1. Precipitation of the crude products from toluene into acetone afforded PFT-Si as a yellow powder in 65% isolated yield, PCT-Si as a pale yellow powder in 74% yield, and PTT-Si as a yellow powder in 57% yield. The number average molecular weights (Mn) were determined by gel permeation chromatography (GPC) in THF against polystyrene standards to be 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500, 6190, and 14700 Da for PFT-Si, PCT-Si, and PTT-Si, respectively, corresponding to an average degree of polymerization of ca. 21, 10, and 25. Treatment of the silylated polymers with a slight excess of BBr3 gave the respective dibromoboryl-substituted polymers. The Si/B exchange reaction was monitored by 1H NMR spectroscopy. For PFT-Si and PCT-Si, the reaction proceeded smoothly and the trimethylsilyl signals in the 1H NMR spectra disappeared completely. However, the reaction of PTT-Si with BBr3 did not go to completion, because of precipitation of the poorly soluble dibromoborylated product. The air-sensitive BBr2-functionalized polymers were directly reacted with 2,4,6-trimethylphenyl copper (MesCu) to replace the bromo substituents with two bulky mesityl groups, leading to greatly improved chemical stability. The boron polymers PFT-B, PCT-B, and PTT-B were obtained as air-stable yellow powdery solids after precipitation into hexanes and freeze-drying from benzene solution. They are thermally stable up to >250 °C according to thermal gravimetric analyses (TGA), although the decomposition temperatures for the boron-functionalized polymers are somewhat lower than those of the silylated precursors (Table 1 and Fig. S1†). The polymers are well soluble in common organic solvents such as THF, chloroform, toluene and dichloromethane, etc., which allowed us to fully characterize their chemical structure and examine their electronic properties in detail.
500, 6190, and 14700 Da for PFT-Si, PCT-Si, and PTT-Si, respectively, corresponding to an average degree of polymerization of ca. 21, 10, and 25. Treatment of the silylated polymers with a slight excess of BBr3 gave the respective dibromoboryl-substituted polymers. The Si/B exchange reaction was monitored by 1H NMR spectroscopy. For PFT-Si and PCT-Si, the reaction proceeded smoothly and the trimethylsilyl signals in the 1H NMR spectra disappeared completely. However, the reaction of PTT-Si with BBr3 did not go to completion, because of precipitation of the poorly soluble dibromoborylated product. The air-sensitive BBr2-functionalized polymers were directly reacted with 2,4,6-trimethylphenyl copper (MesCu) to replace the bromo substituents with two bulky mesityl groups, leading to greatly improved chemical stability. The boron polymers PFT-B, PCT-B, and PTT-B were obtained as air-stable yellow powdery solids after precipitation into hexanes and freeze-drying from benzene solution. They are thermally stable up to >250 °C according to thermal gravimetric analyses (TGA), although the decomposition temperatures for the boron-functionalized polymers are somewhat lower than those of the silylated precursors (Table 1 and Fig. S1†). The polymers are well soluble in common organic solvents such as THF, chloroform, toluene and dichloromethane, etc., which allowed us to fully characterize their chemical structure and examine their electronic properties in detail.
 | ||
| Scheme 1 Synthesis of silylated precursors and borylated copolymers. | ||
| Sample | Yield (%) | M n a/Da | M w a/Da | PDIb | DPnc | T dec d/°C, residuee (%) |
|---|---|---|---|---|---|---|
| a Molecular weight determined by GPC against polystyrene standards in THF. b Polydispersity, PDI = Mw/Mn. c Number average degree of polymerization based on GPC analysis. d Thermal decomposition temperature determined by TGA under nitrogen at 10 °C min−1, based on 5% weight loss. e At 800 °C. | ||||||
| PFT-Si | 65 | 1.35 × 104 | 2.52 × 104 | 1.86 | 21 | 376, 10 |
| PFT-B | 52 | 2.19 × 104 | 4.04 × 104 | 1.85 | 22 | 256, 11 |
| PCT-Si | 74 | 6.19 × 103 | 1.36 × 104 | 2.19 | 10 | 376, 6 |
| PCT-B | 48 | 1.04 × 104 | 2.03 × 104 | 1.95 | 11 | 285, 4 |
| PTT-Si | 57 | 1.47 × 104 | 2.37 × 104 | 1.61 | 25 | 367, 9 |
| PTT-B | 47 | 2.53 × 104 | 5.61 × 104 | 2.22 | 27 | 293, 5 |
Successful functionalization of the conjugated polymers with dimesitylborane acceptor groups was confirmed by NMR spectroscopy. For instance, upon installment of the mesityl groups, a characteristic new set of peaks appeared in the 1H NMR spectra of each of the polymers PFT-B, PCT-B, and PTT-B at around 6.5–6.6 (CH), 2.1–2.2 (Me), and 2.0–2.1 ppm (Me) (Fig. 1). The degree of functionalization is greater than 97% for PFT-B and PCT-B, but for PTT-B integration of the Mes signal at 6.63 ppm (8H) relative to the SiMe3 signal at 0.14 ppm (0.84H) indicates that the conversion for the Si/B exchange reaction is only ca. 95%. A broad resonance in the 11B NMR spectra in the range of 54 to 61 ppm further confirms the formation of the tricoordinate borane species. With an electron-rich arylamine comonomer as the second building block, the 11B NMR signals of PCT-B and PTT-B experience slight upfield shifts to 56 and 54 ppm relative to that of PFT-B at 61 ppm. The number average molecular weights (Mn) measured by GPC range from 10000 to 25000 Da, corresponding to an average of ca. 11 repeating units for PCT-B, 22 for PFT-B, and 27 for PTT-B. Both the degree of polymerization (DPn) and the polydispersity (PDI) are consistent with those of the respective silicon precursors (Table 1).
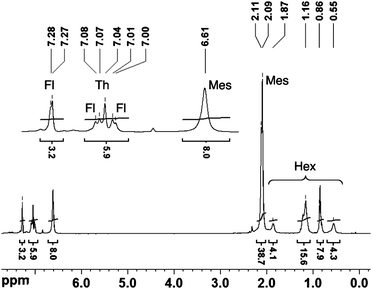 | ||
| Fig. 1 1H NMR spectrum of PFT-B. | ||
The structures of the copolymers PFT-Si, PCT-Si, and PCT-B were further confirmed by MALDI-TOF mass spectroscopy. In all cases, the molecular weights observed by MALDI-TOF MS are significantly lower than those derived from GPC analysis. Given that multiply charged species were not observed, there are two possible reasons: (a) fragmentation of longer chains or (b) the inability to ionize chains with higher mass, such that only smaller chains are seen, which are likely to have hydrogen end groups due to the loss of reactive groups during polymerization (early chain termination). The latter is the most common case for conjugated polymers. For PCT-Si a single set of peaks, nM, with up to 12 repeating units was clearly identified in the mass spectra acquired in (+) ion reflector mode. By comparison with calculated isotope patterns, the peaks for PCT-Si correspond to polymer chains with formula [(Cz-Th2Si2)n]+ (Fig. 2, top). For PCT-B, a corresponding set of peaks, nM, assigned to [(Cz-Th2B2)n]+ was observed, but one extra set of small peaks was found next to each main peak (nM–B in Fig. 2, bottom). The mass difference between these peaks is 249.2 Da corresponding to one BMes2 group; therefore, they are assigned to polymer chains that are missing one BMes2 group. This might be due to the presence of trace amounts of HBr generated during the Si/B exchange reaction, which could lead to a small extent of desilylation of the precursor polymer. Multiple series (four) of peaks are detected for PFT-Si, the two most intense ones are assigned to species [Th2Si2-(Fl-Th2Si2)n-H]+ and [H-(Fl-Th2Si2)n-H]+ (Fig. S2†).12 However, we were not able to obtain a spectrum of sufficient quality for PFT-B.
![MALDI-TOF mass spectra acquired in (+) ion reflector mode for PCT-Si (top, nM = [(Cz-Th2Si2)n]+) and PCT-B (bottom, nM = [(Cz-Th2B2)n]+; nM–B = [(Cz-Th2B2)n–BMes2]+).](/image/article/2011/PY/c0py00343c/c0py00343c-f2.gif) | ||
| Fig. 2 MALDI-TOF mass spectra acquired in (+) ion reflector mode for PCT-Si (top, nM = [(Cz-Th2Si2)n]+) and PCT-B (bottom, nM = [(Cz-Th2B2)n]+; nM–B = [(Cz-Th2B2)n–BMes2]+). | ||
An interesting aspect is the absence of any end groups based on analysis of the MALDI MS peak patterns for PCT-Si and PCT-B. While this could be due to fragmentation of larger polymer chains as mentioned above, an alternative interpretation is that cyclic species are formed preferentially or ionized more efficiently. Considering that the molecular weights of PCT-Si and PCT-B based on GPC analysis are significantly lower than those of the other polymers, the preferred formation of cyclics is certainly possible. This conclusion is further supported by the fact that end groups could not be clearly identified in the 1H NMR spectra.
The photophysical properties of the organoborane polymers and their silylated precursors were examined by UV-Vis absorption and fluorescence spectroscopy. As shown in Fig. 3, for the silyl-substituted polymers a single absorption is found in the region from 300 nm to 450 nm. This band is strongly red-shifted by ca. 70 nm when going to the respective borylated polymers PFT-B, PCT-B, and PTT-B. In addition, a new high-energy band emerges at about 335 nm, independent of the structure of the conjugated main chain skeleton. Therefore, this band is assigned to the 3,3′-bis(dimesitylboryl)-2,2′-bithiophene (Th2B2) chromophore. Based on our previous theoretical calculation on a borylated quaterthiophene species (QT-B), this transition is likely the result of excitation into a cross-conjugation state that involves contributions from the bithiophene moiety and the dimesitylboryl side groups (Fig. 4).
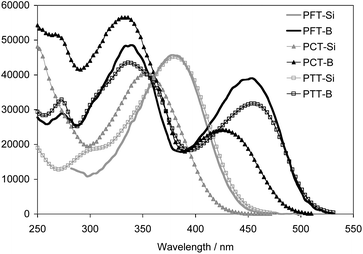 | ||
| Fig. 3 UV-Vis absorption spectra of the polymers in CH2Cl2; the absorptivity is given per polymer repeat unit. | ||
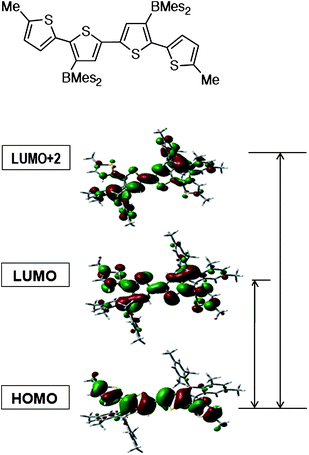 | ||
| Fig. 4 Orbital plots for quaterthiophene QT-B, generated using Gaussview (scaling radii of 75%, isovalue of 0.02, optimization was performed with the Gaussian 03 program using the hybrid density functional B3LYP with the basis set of 6-31G(d)).13 | ||
A more detailed comparison of the silylated and borylated polymers provides further insight into the effect of the electron-deficient borane moieties and the role of the electronic structure of the comonomer. The fluorene and triarylamine-containing polymers PFT-Si and PTT-Si exhibit quite similar absorption maxima at around 380 nm. However, the absorptions are red-shifted relative to that of PCT-Si at 355 nm. Similarly, the low energy absorption bands for PFT-B and PTT-B centered at around 454 nm are red-shifted by about 32 nm in comparison to that for PCT-B. A reason could be that the 3,6-linkage of the carbazole moieties leads to only moderate delocalization throughout the polymer main chain.10,14 We estimated the optical band gaps of the boron polymers from the absorption onsets to be 2.45 eV for PFT-B, 2.55 eV for PCT-B, and 2.42 eV for PTT-B. They are by about 0.4 eV smaller than those of the silylated polymers, reflecting the effect of functionalization with dimesitylboryl groups (Table 2).
| Sample | E 0′Reda/mV | E 0′Oxb/mV | λ abs/nm (ε/M−1 cm−1)c | λ em/nm (λexc/nm)c | Optical gapd/eV | Φ fl e | λ abs f/nm | λ em f , g/nm |
|---|---|---|---|---|---|---|---|---|
| a From cyclic voltammetry in THF/0.1 M Bu4N[PF6], 100 mV s−1, referenced vs. Fc/Fc+ couple. b From square wave voltammograms in CH2Cl2/0.1 M Bu4N[PF6], 100 mV s−1, referenced vs. Fc/Fc+ couple; data in brackets are from cyclic voltammetry. c In CH2Cl2. d Calculated from the absorption onset. e Referenced vs.diphenylanthracene. f Thin film. g Excited at λabs,max. | ||||||||
| PFT-Si | −2480, −2710 (ir) | 600 (620), 960 (nr) | 380 (4.58 × 104) | 484 (380) | 2.84 | 0.19 | 379 | 481 |
| PFT-B | −2260, −2440 | 560 (620), 940 (nr) | 453 (3.96 × 104), 337 (4.85 × 104) | 533 (453) | 2.45 | 0.07 | 443, 340 | 536 |
| PCT-Si | −2640, −2880 | 500 (nr), 580 (570) | 355 (4.05 × 104) | 468 (355) | 2.96 | 0.10 | 357 | 487 |
| PCT-B | −2360, −2530 | 460 (470), 620 (610), 830 (830) | 422 (2.49 × 104), 334 (5.68 × 104) | 527 (422) | 2.55 | 0.02 | 431, 344 | 509 |
| PTT-Si | −2650, −2990 | 380 (380), 700 (710), 930 (930) | 381 (4.52 × 104) | 493 (381) | 2.79 | 0.14 | 376 | 488 |
| PTT-B | −2340, −2520 | 390 (380), 750 (750), 940 (970) | 455 (3.17 × 104), 337 (4.35 × 104) | 546 (455) | 2.42 | 0.04 | 445, 340 | 548 |
A comparison of these results with data for the all-thiophene polymers PT-Si and PT-B7 reveals that the lowest energy absorptions for the carbazole polymers PCT-Si and PCT-B are similar to those of silylated PT-Si and borylated PT-B, respectively, whereas the transitions for the fluorene and triarylamine polymers are considerably lower in energy.
All polymers are fluorescent and their emission spectra are illustrated in Fig. 5. The silylated polymers emit in the blue green region with maxima ranging from 468 to 493 nm. Upon borylation, a single emission band emerges in the green region with a maximum at 533 nm for PFT-B, 527 nm for PCT-B, and 546 nm for PTT-B. These bands are red shifted by about 50 nm relative to those of the silylated polymers, which again is attributed to the electronic effect of the Mes2B π-acceptor functionalities. However, in contrast to what we found from the absorption spectra, all the copolymers emit at much shorter wavelength than the thiophene polymers PT-Si (554 nm) and PT-B (617 nm).7 A possible reason may be that electronic delocalization in the excited state is more pronounced for the polythiophenes than the copolymers with fluorene, carbazole, or triarylamine moieties. The quantum efficiencies for the silylated copolymers range from 10% to 20%. Those of the borylated polymers are comparatively lower, which is likely in part related to the lower optical gap for the organoboron polymers, which favors non-radiative decay.
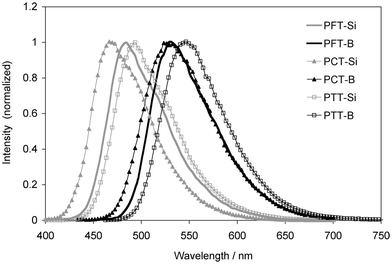 | ||
| Fig. 5 Normalized fluorescence spectra of the polymers in CH2Cl2. | ||
Variations in the polarity of the organic solvent (CH2Cl2, THF, pyridine) led to only minor differences in the absorption and emission profiles and thus no marked solvatochromic effect was observed (Fig. S3 and S4†). Moreover, thin films exhibited absorption and emission profiles similar to the ones in solution (Table 2, Fig. S5 and S6†), indicating that inter-chain aggregation and excimer formation are not favorable for these types of polymers. Presumably, the bulky dimesitylboryl substituents prevent alignment of the conjugated polymer chains.
The electrochemical properties were studied by cyclic voltammetry and square wave voltammetry (Fig. 6). Two quasi-reversible cathodic waves were observed for each of the boron polymers. They are assigned to successive reduction of the two boron centers connected by the bithiophene moieties. The difference in the redox potential for the two waves, ΔE, is about 180 mV, which is slightly smaller than the peak separation of 230 mV found for PT-B.7 The first reduction occurs at significantly less negative potential than that for the silicon polymers (ΔE > 200 mV), which further illustrates that the LUMO energy level is lowered by attachment of the electron withdrawing Mes2B functionalities. The LUMO energy levels are also affected by the nature of the conjugated main chain skeleton, although the effect is less pronounced. Due to the relatively more electron-rich arylamine comonomers, PCT-B and PTT-B are reduced at more negative potentials than PFT-B. This trend also holds for polymers PFT-Si, PCT-Si and PTT-Si.
![Cyclic voltammograms of (top) PFT-Si and PFT-B, (middle) PCT-Si and PCT-B, (bottom) PTT-Si and PTT-B; anodic scan: CH2Cl2/0.1 M Bu4N[PF6], 100 mV s−1; cathodic scan: THF/0.1 M Bu4N[PF6], 100 mV s−1; reported relative to the Fc/Fc+ couple.](/image/article/2011/PY/c0py00343c/c0py00343c-f6.gif) | ||
| Fig. 6 Cyclic voltammograms of (top) PFT-Si and PFT-B, (middle) PCT-Si and PCT-B, (bottom) PTT-Si and PTT-B; anodic scan: CH2Cl2/0.1 M Bu4N[PF6], 100 mV s−1; cathodic scan: THF/0.1 M Bu4N[PF6], 100 mV s−1; reported relative to the Fc/Fc+ couple. | ||
In contrast to our observations for the reduction processes, polymer oxidation is not strongly influenced by the nature of the side chain substituents. The potential differences between the boron polymers and the silicon precursors are less than 50 mV. For instance, a reversible oxidation wave is found at 380 mV for PTT-Si and shifted to 390 mV for PTT-B. This redox wave is accompanied by two additional less well-resolved oxidation waves at higher potentials of ca. 700–750 mV and 930–950 mV, respectively. In the case of PCT-Si and PCT-B similar oxidation waves are observed, but the initial redox process occurs at higher potential leading to a series of overlapping and therefore less resolved oxidation events, while for PFT-Si and PFT-B only the redox processes at higher potential are observed. Based on these observations we assign the less anodic processes in PTT-Si, PTT-B, PCT-Si, and PCT-B to the arylamine moieties, while the second feature that is also found for PFT-Si and PFT-B is attributed to oxidation of the conjugated polymer main chain. The assignment of the first oxidation process to the arylamine moieties was further confirmed by a spectroelectrochemical study on PTT-B, which shows well-separated oxidation processes at 390, 750 and 940 mV in its square wave voltammogram (Table 2). Upon preparative oxidation of PTT-B in an OTTLE (Optical Transparent Thin-Layer Electrochemical) cell15 under N2 protection, the emergence of two new absorption bands at ca. 660 nm and 570 nm (shoulder) was observed (Fig. 7). This is a characteristic feature of oxidation of the triarylamine groups according to prior literature reports16 on UV-visible spectra of radical cations derived from differently substituted triphenylamine derivatives.
![UV-Vis spectra of PTT-B before and after preparative oxidation in CH2Cl2 containing 0.1 M Bu4N[PF6].](/image/article/2011/PY/c0py00343c/c0py00343c-f7.gif) | ||
| Fig. 7 UV-Vis spectra of PTT-B before and after preparative oxidation in CH2Cl2 containing 0.1 M Bu4N[PF6]. | ||
Conclusions
A series of conjugated copolymers that feature trimethylsilyl or electron-withdrawing dimesitylboryl groups as side chains have been prepared. The structure of the polymer main chain was systematically varied by Suzuki-type coupling of diiodinated 4,4′-bis(trimethylsilyl)-2,2′-bithiophene with different conjugated comonomers. Dimesitylboryl functionalities were then introduced by a Si/B exchange reaction with BBr3, followed by nucleophilic substitution of bromine for sterically hindered mesityl groups. The resulting polymeric materials are well soluble in common organic solvents, exhibit high thermal stability and are stable in air. Their electronic structure was examined in detail by cyclic voltammetry, UV-Vis and fluorescence spectroscopy. These studies show that upon going from the silicon polymers to the respective boron polymers the HOMO–LUMO energy gap becomes smaller. This results from lowering of the LUMO energy level by modification with electron accepting dimesitylboryl groups, while the HOMO energy level remains largely unaffected. On the other hand, variation of the conjugated main chain by using different organic π systems (fluorene, carbazole, triphenylamine) as building blocks affects both the HOMO and the LUMO energy levels. According to cyclic voltammetry studies, both the HOMO and LUMO levels for PCT-B and PTT-B are higher in energy in comparison to those of PFT-B. We attribute this to the electron-rich nature of the arylamine building block embedded in the main chain of PCT-B and PTT-B. From comparison of the absorption and emission data, subtle differences in the HOMO and LUMO energies lead to a slightly higher energy gap for polymers PCT-Si and PCT-B in comparison to the other silicon and boron-modified conjugated polymers.Experimental section
Materials and methods
Fluorene, carbazole, diphenylamine, and BBr3 were purchased from Aldrich, nBuLi (1.6 M in hexanes) from Fisher Scientific, and Me3SnCl from Strem Chemicals. N,N-Bis(4-bromophenyl)-2,4,6-trimethylaniline,17mesitylcopper (MesCu),182,7-dibromo-9,9′-dihexylfluorene,82,2′-(9,9-dihexyl-fluorene-2,7-diyl)-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) (Fl(Bpin)2),199-(4-tert-butylphenyl)-3,6-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)carbazole (Cz(Bpin)2),192,4,6-trimethyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (TPA(Bpin)2),19 and 5,5′-diiodo-4,4′-bis(trimethylsilyl)-2,2′-bithiophene (Th2Si2I2)7 were prepared according to literature procedures. All air or moisture-sensitive reactions and manipulations were carried out under an atmosphere of pre-purified nitrogen using Schlenk techniques or an inert atmosphere glove box (mBraun). Ether solvents were distilled from Na/benzophenone prior to use. Hydrocarbon and chlorinated solvents were purified using a solvent purification system (Innovative Technologies, alumina/copper columns for hydrocarbon solvents), and chlorinated solvents were subsequently distilled from CaH2 and degassed via several freeze–pump–thaw cycles.The 499.9 MHz 1H and 125.7 MHz 13C NMR spectra were recorded on a Varian INOVA 500 MHz spectrometer. The 160.4 MHz 11B NMR spectra were recorded with a boron-free probe using boron-free quartz NMR tubes. The 1H and 13C NMR spectra were referenced internally to the solvent peaks and the 11B NMR spectra externally to BF3·Et2O (δ = 0) in C6D6. UV-visible absorption data were acquired on a Varian Cary 500 UV-Vis/NIR spectrophotometer. The fluorescence data and quantum yields were measured on a Varian Cary Eclipse fluorescence spectrophotometer with optically dilute solutions (A < 0.1). Diphenylanthracene20 was used as the standard for determination of the quantum yields (Φ). Cyclic voltammetry was carried out on a CV-50W analyzer from BAS. The three-electrode system consisted of an Au disk as a working electrode, a Pt wire as a secondary electrode and a Ag wire as the pseudo-reference electrode. The voltammograms were recorded with ca. 10−3 to 10−4 M solutions in THF (reduction waves) or CH2Cl2 (oxidation waves) containing Bu4N[PF6] (0.1 M) as the supporting electrolyte. The scans were referenced after the addition of a small amount of ferrocene (Fc, reduction cycles) or decamethylferrocene (Fc*, oxidation cycles) as internal standard. The potentials are reported relative to the Fc/Fc+ couple (Fc*/Fc*+ = −548 mV vs.Fc/Fc+ in CH2Cl2/Bu4N[PF6]). Spectroelectrochemistry was performed using a home-built OTTLE (Optical Transparent Thin-Layer Electrochemical) cell,15 a Perkin Elmer Lambda 16 UV/VIS spectrometer, and an EG&G Princeton Applied Research 263A potentiostat. The measurement was recorded on a ca. 10−4 M solution in CH2Cl2 containing Bu4N[PF6] (0.1 M) as the supporting electrolyte.
GPC analyses were performed in THF (1 mL min−1) using a Waters Empower system equipped with a 717plus autosampler, a 1525 binary HPLC pump, a 2998 Photodiode Array Detector, and a 2414 refractive index detector (880 nm). Three styragel columns (Polymer Laboratories; two PLgel 5 μm Mixed-C and one PLgel 5 μm Mixed-D), which were kept in a column heater at 35 °C, were used for separation. The columns were calibrated with narrow polystyrene standards (Polymer Laboratories). MALDI-TOF measurements were performed on an Applied Biosystems 4800 Proteomics Analyzer in reflectron (+) mode with delayed extraction. Benzo[a]pyrene was used as the matrix (10 mg mL−1 in toluene). Thermogravimetric analysis (TGA) measurement was performed on a Perkin Elmer Pyris 1 Thermogravimetric Analyzer at a scan rate of 10 °C min−1 and up to 800 °C.
Preparation of PFT-Si
An equimolar quantity of Th2Si2I2 (0.96 g, 1.71 mmol) and Fl(Bpin)2 (1.00 g, 1.71 mmol) was dissolved in 5 mL of toluene, Pd(PPh3)4 (39.6 mg, 0.034 mmol) was added and the mixture was transferred into a reaction tube, followed by addition of 4.5 mL of a degassed solution of aqueous K2CO3 (2.0 M). The reaction mixture was slowly heated to 110 °C and kept stirring at that temperature for 2 days. The mixture was then cooled back to room temperature and the catalyst residue was removed by passing the organic layer through a short plug of alumina. The yellow filtrate was concentrated to 2 mL and precipitated into 150 mL of acetone. The product was isolated as yellow powder and dried under high vacuum at 50 °C. Yield: 0.70 g, 65%. 1H NMR (499.9 MHz, CDCl3): δ = 7.76 (d, 3J = 8.5 Hz, 2H, Fl), 7.49 (m, 4H, Fl), 7.22 (s, 2H, Th), 2.05 (br, 4H, Hex), 1.2–1.0 (m, 12H, Hex), 0.79 (t, 3J = 7.5 Hz, 6H, Hex), 0.68 (br, 4H, Hex), 0.20 ppm (s, 18H, SiMe3); 13C NMR (125.7 MHz, CDCl3): δ = 151.2, 141.0, 139.0, 136.7, 135.4, 130.8, 129.0, 124.4, 119.7 (aromatic C), 55.7 (Fl), 41.0, 31.8, 29.9, 24.0, 22.8, 14.2 (hexyl), 0.8 (SiMe3) ppm; 29Si NMR (99.3 MHz, CDCl3): δ = −8.1 ppm; GPC-RI (in THF against polystyrene standards) Mn = 1.35 × 104, Mw = 2.52 × 104, PDI = 1.86; UV-Vis (CH2Cl2, 3.39 × 10−6 M): λmax (ε, M−1 cm−1) = 380 nm (4.58 × 104); fluorescence (CH2Cl2, 3.39 × 10−6 M): λem,max = 484 nm (λexc = 380 nm, Φ = 18.6%); TGA (N2, 10 °C min−1): Tdec = 376 °C (onset), 10% residue at 800 °C.Preparation of PFT-B
At −35 °C, a solution of PFT-Si (200 mg, 0.31 mmol) in 4 mL of CH2Cl2 was added dropwise to a solution of BBr3 (194 mg, 0.77 mmol) in 2 mL of CH2Cl2. A green solution formed, which was gradually warmed up to room temperature and kept stirring overnight. The volatile materials were evaporated under high vacuum. Without further purification, the crude product was re-dissolved in 5 mL of toluene and mixed with a solution of MesCu (273 mg, 1.50 mmol) in 5 mL of toluene. The reaction mixture was slowly heated to 110 °C and kept stirring at that temperature for 2 days. After cooling back to room temperature, the solid by-product (CuBr) was removed by passing the mixture through a fritted glass funnel, followed by a short plug of alumina. The filtrate was concentrated to less than 1 mL and precipitated into 50 mL of cold hexanes. The final product was obtained by freeze-drying from benzene as a yellow solid. Yield: 160 mg, 52%. 1H NMR (499.9 MHz, CDCl3): δ = 7.28 (s, 2H, Fl), 7.07 (d, 3J = 7.5 Hz, 2H, Fl), 7.04 (s, 2H, Th), 7.01 (d, 3J = 7.5 Hz, 2H, Fl), 6.61 (br, 8H, Mes), 2.12 (s, 12H, p-Me), 2.09 (s, 24H, o-Me), 1.86 (br, 4H, Hex), 1.3–1.0 (m, 12H, Hex), 0.85 (t, 6H, 3J = 7.0 Hz, Hex), 0.56 ppm (br, 4H, Hex); 13C NMR (125.7 MHz, CDCl3): δ = 155.6, 150.8, 147.4 (Fl/Th), 142.5 (Mes), 140.7 (Mes), 140.2 (Fl/Th), 138.9 (Mes), 136.0, 134.0, 131.8, 129.4 (Fl/Th), 128.4 (Mes), 122.5, 118.2 (Fl/Th), 55.1 (Fl), 40.2, 31.7, 29.9 (Hex), 23.4 (o-Me and p-Me), 22.9, 21.3, 14.3 (Hex) ppm; 11B NMR (160.3 MHz, CDCl3): δ = 61 ppm (w1/2 = 6240 Hz); GPC-RI (in THF against PS standards) Mn = 2.19 × 104, Mw = 4.04 × 104, PDI = 1.85; UV-Vis (CH2Cl2, 1.57 × 10−6 M): λmax (ε, M−1 cm−1) = 453 nm (3.96 × 104), 337 nm (4.85 × 104); fluorescence (CH2Cl2, 1.57 × 10−6 M): λem,max = 533 nm (λexc = 453 nm, Φ = 7.3%); TGA (N2, 10 °C min−1): Tdec = 256 °C (onset), 11% residue at 800 °C. Elemental analysis calcd for [C69H78B2S2]n C 83.45, H 7.92; found C 81.70, H 7.80%.Preparation of PCT-Si
Polymer PCT-Si was prepared in the same manner as described for PFT-Si by using Cz(Bpin)2 (491 mg, 0.89 mmol), Th2Si2I2 (500 mg, 0.89 mmol), Pd(PPh3)4 (21.0 mg, 0.018 mmol) and 2.5 mL of aqueous K2CO3 solution (2.0 M). The product was isolated as a pale yellow powder. Yield: 400 mg, 74%. 1H NMR (499.9 MHz, CDCl3): δ = 8.25 (s, 2H, Cz), 7.65 (d, 3J = 8.5 Hz, 2H, Cz), 7.56 (m, 4H, Ph), 7.46 (d, 3J = 8.5 Hz, 2H, Cz), 7.22 (s, 2H, Th), 1.45 (s, 9H, t-Bu), 0.19 ppm (s, 18H, SiMe3); 13C NMR (125.7 MHz, CDCl3): δ = 151.2, 151.0, 141.5, 138.7, 136.7, 134.8, 130.7, 128.4, 128.3, 127.1, 126.7, 123.1, 121.8, 109.9 (aromatic C), 35.1, 31.7 (t-Bu), 0.9 (SiMe3) ppm; 29Si NMR (99.3 MHz, CDCl3): δ = −7.8 ppm; GPC-RI (in THF against PS standards) Mn = 6.19 × 103, Mw = 1.36 × 104, PDI = 2.19; UV-Vis (CH2Cl2, 1.30 × 10−5 M): λmax (ε, M−1 cm−1) = 355 nm (4.05 × 104); fluorescence (CH2Cl2, 1.30 × 10−5 M): λem,max = 468 nm (λexc = 355 nm, Φ = 10.1% relative to DPA); TGA (N2, 10 °C min−1): Tdec = 376 °C (onset), 6% residue at 800 °C.Preparation of PCT-B
Polymer PCT-B was prepared in the same manner as described for PFT-B using PCT-Si (100 mg, 0.17 mmol), BBr3 (107 mg, 0.42 mmol) and MesCu (147 mg, 0.81 mmol). The product was obtained as yellow powder. Yield: 76 mg, 48%. 1H NMR (499.9 MHz, CDCl3): δ = 7.76 (s, 2H, Cz), 7.60 (d, 3J = 8.0 Hz, 2H, Ph), 7.31 (d, 3J = 8.5 Hz, 2H, Cz), 7.26 (d, 3J = 8.0 Hz, 2H, Ph), 7.09 (s, 2H, Th), 7.00 (d, 3J = 8.5 Hz, Cz), 6.49 (br s, 8H, Mes), 2.13 (br, 24H, o-Me), 1.96 (br, 12H, p-Me), 1.44 ppm (s, 9H, t-Bu); 13C NMR (125.7 MHz, CDCl3): δ = 156.6, 150.6, 146.9 (Cz/Th), 142.4 (Mes), 140.8 (Cz/Th), 140.5 (Mes), 138.3 (Mes), 135.7, 135.0, 131.8 (Cz/Th), 128.3 (Mes), 127.7, 127.1, 127.0, 126.2, 122.8, 121.6, 108.6 (Cz/Th), 35.0, 31.7 (t-Bu), 23.5 (o-Me), 21.4 (p-Me) ppm; 11B NMR (160.3 MHz, CDCl3): δ = 56 ppm (w1/2 = 8000 Hz); GPC-RI (in THF against PS standards) Mn = 1.04 × 104, Mw = 2.03 × 104, PDI = 1.95; UV-Vis (CH2Cl2, 1.73 × 10−6 M): λmax (ε, M−1 cm−1) = 422 (2.49 × 104), 334 nm (5.68 × 104); fluorescence (CH2Cl2, 1.73 × 10−6 M): λem,max = 527 nm (λexc = 422 nm, Φ = 2.0%); TGA (N2, 10 °C min−1): Tdec = 285 °C (onset), 4% residue at 800 °C. Elemental analysis calcd for [C66H65B2NS2]n C 82.75, H 6.84, N 1.46; found C 80.24, H 6.56, N 1.22%.Preparation of PTT-Si
Polymer PTT-Si was prepared in the same manner as described for PFT-Si by using Th2Si2I2 (500 mg, 0.89 mmol), TPA(Bpin)2 (481 mg, 0.89 mmol), Pd(PPh3)4 (7.4 mg, 0.0064 mmol) and 2.5 mL of aqueous solution of K2CO3 (2.0 M). The product was isolated as yellow powder. Yield: 302 mg, 57%. 1H NMR (499.9 MHz, CDCl3): δ = 7.31 (d, 3J = 8.5 Hz, 4H, Ph), 7.12 (s, 2H, Th), 7.02 (d, 3J = 8.5 Hz, 4H, Ph), 6.99 (s, 2H, Mes), 2.36 (s, 3H, p-Me), 2.05 (s, 6H, o-Me), 1.34 (s, 0.82H, Bpin end group), 0.15 ppm (s, 18H, SiMe3); 13C NMR (125.7 MHz, CDCl3): δ = 150.4, 146.0, 139.9, 138.5, 137.7, 137.3, 136.3, 130.8, 130.5, 130.3, 128.9, 119.3 (aromatic C), 25.1 (Bpin end group), 23.8 (Bpin end group), 21.3 (p-Me), 18.8 (o-Me), 0.7 (SiMe3) ppm; 29Si NMR (99.3 MHz, CDCl3): δ = −8.1 ppm; GPC-RI (in THF against PS standards) Mn = 1.47 × 104, Mw = 2.37 × 104, PDI = 1.61; UV-Vis (CH2Cl2, 3.03 × 10−5 M): λmax (ε, M−1 cm−1) = 381 nm (4.52 × 104); fluorescence (CH2Cl2, 3.03 × 10−5 M): λem,max = 493 nm (λexc = 381 nm, Φ = 13.2%); TGA (N2, 10 °C min−1): Tdec = 367 °C (onset), 9% residue at 800 °C.Preparation of PTT-B
At −35 °C, a solution of PTT-Si (100 mg, 0.17 mmol) in 10 mL of toluene was added to a solution of BBr3 (95 mg, 0.38 mmol) in 2 mL of CH2Cl2. A dark green solution formed, which was gradually warmed up to room temperature and kept stirring for 24 h. The mixture was concentrated under high vacuum, such that a small amount of solvent remained (it is difficult to re-dissolve the compound when it is fully dried). After adding more toluene (2 mL), the intermediate was mixed with a solution of MesCu (140 mg, 0.77 mmol) in 5 mL of toluene. The reaction mixture was heated to 110 °C and kept stirring for 2 days. After cooling to room temperature, the solid by-product (CuBr) was removed by passing the mixture through a fritted glass funnel, followed by a short plug of alumina. The filtrate was concentrated to less than 1 mL and precipitated into 50 mL of cold hexanes. The final product was obtained by freeze-drying from benzene as a yellow solid. Yield: 76 mg, 47%. 1H NMR (499.9 MHz, CDCl3): δ = 7.01 (d, 3J = 8.0 Hz, 4H, Ph), 6.97 (s, 2H, Th), 6.89 (s, 2H, Mes-N), 6.63 (s, 8H, Mes-B), 6.42 (d, 3J = 8.5 Hz, Ph), 2.32 (s, 3H, NMes p-Me), 2.20 (s, 12H, BMes p-Me), 2.05 (s, 24H, BMes o-Me), 1.78 (s, 6H, NMes o-Me), 0.14 ppm (0.84H, 5% residual SiMe3); 13C NMR (125.7 MHz, CDCl3): δ = 155.5, 147.4, 145.6 (aromatic C), 142.5 (BMes), 140.7 (BMes), 139.5 (aromatic C), 138.4 (BMes), 137.7, 137.2, 135.6, 131.2, 130.0 (aromatic C), 128.3 (BMes), 128.1, 118.4 (aromatic C), 23.4 (BMes o-Me), 21.5 (BMes p-Me), 21.3 (NMes p-Me), 18.4 (NMes o-Me), 0.66 (residual SiMe3) ppm; 11B NMR (160.3 MHz, CDCl3): δ = 54 ppm (w1/2 = 6400 Hz); GPC-RI (in THF against PS standards) Mn = 2.53 × 104, Mw = 5.61 × 104, PDI = 2.22; UV-Vis (CH2Cl2, 1.57 × 10−6 M): λmax (ε, M−1 cm−1) = 455 (3.17 × 104), 337 (4.35 × 104), 273 nm (3.28 × 104); fluorescence (CH2Cl2, 1.78 × 10−5 M): λem,max = 546 nm (λexc = 455 nm, Φ = 4.1%); TGA (N2, 10 °C min−1): Tdec = 293 °C (onset), 5% residue at 800 °C. Elemental analysis calcd for [C65H65B2NS2]n C 82.53, H 6.93, N 1.48; found C 81.29, H 7.05, N 1.35%.Acknowledgements
We thank the National Science Foundation (CHE-0809642) for financial support. FJ thanks the Alfred P. Sloan Foundation for a research fellowship and the Alexander-von-Humboldt Foundation for a Friedrich Wilhelm Bessel Research Award. We thank Frauke Schödel and Prof. Dr M. Wagner at the Johann-Wolfgang-Goethe Universität Frankfurt for kindly performing the spectroelectrochemical measurement and Jose Medina for his efforts in preparing compound QT-SiMe3.Notes and references
- (a) Special issue on Organic Electronics and Optoelectronics in Chem. Rev., 2007, 107, 923–1386 Search PubMed; (b) Special issue on Organic Photovoltaics in Acc. Chem. Res., 2009, 42, 1689–1857 Search PubMed; (c) S. W. Thomas, III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS.
- (a) Handbook of Oligo- and Polythiophenes, ed. D. Fichou, Wiley-VCH, Weinheim, 1999 Search PubMed; (b) N. Somanathan and S. Radhakrishnan, Int. J. Mod. Phys. B, 2005, 19, 4645–4676 CrossRef CAS; (c) I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS; (d) I. F. Perepichka and D. F. Perepichka, Handbook of Thiophene-Based Materials, 2009, vol. 1, pp. 341–364 Search PubMed.
- (a) M. O. Wolf, Adv. Mater., 2001, 13, 545–553 CrossRef CAS; (b) A. S. Abd-El-Aziz, C. E. Carraher, Jr, C. U. Pittman, Jr, J. E. Sheats and M. Zeldin, Macromolecules Containing Metal and Metal-Like Elements: A Half-Century of Metal- and Metalloid-Containing Polymers, John Wiley & Sons, Hoboken, 2003, vol. 1 Search PubMed; (c) I. Manners, Synthetic Metal-Containing Polymers, Wiley-VCH, 2004 Search PubMed; (d) R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2004, 126, 2268–2269 CrossRef CAS; (e) T. Baumgartner and R. Reau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS; (f) M. J. Maclachlan, in Frontiers in Transition Metal-Containing Polymers, ed. A. S. Abd-El-Aziz and I. Manners, John Wiley & Sons, Inc., Hoboken, NJ, 2007, pp. 161–215 Search PubMed; (g) K. Naka, Polym. J., 2008, 40, 1031–1041 CrossRef CAS; (h) Z. Jin and B. L. Lucht, J. Am. Chem. Soc., 2005, 127, 5586–5595 CrossRef CAS; (i) N. Mukherjee and R. M. Peetz, Macromolecules, 2008, 41, 6677–6685 CrossRef CAS; (j) J. Mei, K. Ogawa, Y. G. Kim, N. C. Heston, D. J. Arenas, Z. Nasrollahi, T. D. McCarley, D. B. Tanner, J. R. Reynolds and K. S. Schanze, ACS Appl. Mater. Interfaces, 2009, 1, 150–161 CrossRef CAS; (k) L. V. Interrante and J. S. Rathore, Dalton Trans., 2010, 39, 9193–9202 RSC.
- (a) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS; (b) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS; (c) N. Matsumi and Y. Chujo, Polym. J., 2008, 40, 77–89 CrossRef CAS; (d) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef CAS.
- (a) N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 5112–5113 CrossRef CAS; (b) N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 10776–10777 CrossRef CAS; (c) Y. Chujo, Y. Sasaki, N. Kinomura and N. Matsumi, Polymer, 2000, 41, 5047–5051 CrossRef CAS; (d) A. Nagai, T. Murakami, Y. Nagata, K. Kokado and Y. Chujo, Macromolecules, 2009, 42, 7217–7220 CrossRef CAS; (e) J. B. Heilmann, M. Scheibitz, Y. Qin, A. Sundararaman, F. Jäkle, T. Kretz, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2006, 45, 920–925 CrossRef CAS; (f) J. B. Heilmann, Y. Qin, F. Jäkle, H. W. Lerner and M. Wagner, Inorg. Chim. Acta, 2006, 359, 4802–4806 CrossRef CAS; (g) A. Lorbach, M. Bolte, H. Li, H.-W. Lerner, M. C. Holthausen, F. Jäkle and M. Wagner, Angew. Chem., Int. Ed., 2009, 48, 4584–4588 CrossRef CAS; (h) M. Scheibitz, H. Li, J. Schnorr, A. Sánchez Perucha, M. Bolte, H.-W. Lerner, F. Jäkle and M. Wagner, J. Am. Chem. Soc., 2009, 131, 16319–16329 CrossRef CAS; (i) W. J. Niu, M. D. Smith and J. J. Lavigne, J. Am. Chem. Soc., 2006, 128, 16466–16467 CrossRef; (j) J. Chai, C. Wang, L. Jia, Y. Pang, M. Graham and S. Z. D. Cheng, Synth. Met., 2009, 159, 1443–1449 CrossRef CAS; (k) I. Yamaguchi, T. Tominaga and M. Sato, Polym. Int., 2009, 58, 17–21 CrossRef CAS.
- (a) A. Sundararaman, M. Victor, R. Varughese and F. Jäkle, J. Am. Chem. Soc., 2005, 127, 13748–13749 CrossRef CAS; (b) H. Li and F. Jäkle, Macromol. Rapid Commun., 2010, 31, 915–920 CrossRef CAS; (c) V. D. B. Bonifácio, J. Morgado and U. Scherf, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2878–2883 CrossRef CAS; (d) W. Liu, M. Pink and D. Lee, J. Am. Chem. Soc., 2009, 131, 8703–8707 CrossRef CAS.
- H. Li, A. Sundararaman, K. Venkatasubbaiah and F. Jäkle, J. Am. Chem. Soc., 2007, 129, 5792–5793 CrossRef CAS.
- H. Li and F. Jäkle, Angew. Chem., Int. Ed., 2009, 48, 2313–2316 CAS.
- C. H. Zhao, A. Wakamiya and S. Yamaguchi, Macromolecules, 2007, 40, 3898–3900 CrossRef CAS.
- D. Reitzenstein and C. Lambert, Macromolecules, 2009, 42, 773–782 CrossRef CAS.
- (a) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CrossRef CAS; (b) F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef; (c) K. Parab, K. Venkatasubbaiah and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 12879–12885 CrossRef CAS; (d) T. W. Hudnall, C.-W. Chiu and F. P. Gabbai, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS; (e) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS; (f) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbai, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS; (g) M. Miyata and Y. Chujo, Polym. J., 2002, 34, 967–969 CrossRef CAS; (h) K. Parab and F. Jäkle, Macromolecules, 2009, 42, 4002–4007 CrossRef CAS.
- The presence of multiple different end groups for polymers PFT-Si and PFT-B prevented an estimation of the molecular weight by 1H NMR end group analysis.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- The respective polymers with 2,7-carbazole linkers might be expected to show considerably longer wavelength absorptions as a result of more extensive π-conjugation throughout the polymer backbone. See: P.-L. T. Boudreault, S. Beaupre and M. Leclerc, Polym. Chem., 2010, 1, 127–136 Search PubMed.
- M. Krejčik, M. Daněk and F. Hartl, J. Electroanal. Chem., 1991, 317, 179–187 CrossRef CAS.
- S. Amthor, B. Noller and C. Lambert, Chem. Phys., 2005, 316, 141–152 CrossRef CAS.
- M. W. Hooper, M. Utsunomiya and J. F. Hartwig, J. Org. Chem., 2003, 68, 2861 CrossRef CAS.
- (a) T. Tsuda, T. Yazawa, K. Watanabe, T. Fujii and T. Saegusa, J. Org. Chem., 1981, 46, 192–194 CrossRef CAS; (b) A. Sundararaman and F. Jäkle, J. Organomet. Chem., 2003, 681, 134–142 CrossRef CAS.
- M. Ranger, D. Rondeau and M. Leclerc, Macromolecules, 1997, 30, 7686–7691 CrossRef CAS.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, Marcel Dekker Inc., New York, 1993 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA plots for all polymers, MALDI-TOF mass spectrum for PFT-Si, absorption and emission spectra in different solvents and of polymer thin films. See DOI: 10.1039/c0py00343c |
| This journal is © The Royal Society of Chemistry 2011 |