Nitrones in synthetic polymer chemistry
Edgar H. H.
Wong
ab,
Tanja
Junkers
*c and
Christopher
Barner-Kowollik
*a
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128, Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; Fax: +49 721-608-45740; Tel: +49 721-608-45641
bCentre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
cInstitute for Materials Research, Polymer Reaction Design Group, Universiteit Hasselt, Universiteit Hasselt, Agoralaan, Gebouw D, B-3590, Diepenbeek, Belgium. E-mail: tanja.junkers@uhasselt.be; Fax: +32 1126-8399; Tel: +32 1126-8318
First published on 24th January 2011
Abstract
Nitrones are arguably one of the most efficient compounds with multi-functional capabilities, acting as both radical spin traps and 1,3-dipoles. In contrast to their potential synthetic versatility, the application of nitrones in synthetic polymer chemistry is still in its infancy and its true potential has yet to be fully explored. The current report thus serves as a highlight and provides an update on the current status of nitrones in macromolecular synthesis.
 Edgar H. H. Wong | Edgar H. H. Wong obtained his BE in Chemical Engineering (Honours Class I) in December 2007 from the University of New South Wales, Australia. Soon thereafter, he commenced a PhD under the joint supervision of Christopher Barner-Kowollik, Thomas Junkers and Martina Stenzel—developing new macromolecular engineering protocols using nitrone radical chemistry. He will graduate with a PhD in 2011 and continue research in the field of polymer science. His other main research interests include photoinitiated polymerization systems, novel materials synthesis, efficient polymer ligation chemistries as well as polymer reaction kinetics and mechanisms. |
 Tanja Junkers | Tanja Junkers graduated in 2006 from the University of Göttingen with a PhD on radical polymerization kinetics. Subsequently, she joined the Centre for Advanced Macromolecular Design at the University of New South Wales in Sydney where she worked on new control methods for radical reactions. In 2008 she started a habilitation in macromolecular chemistry at the Karlsruhe Institute of Technology. Early in 2010 she was appointed Professor at Hasselt University where she founded the Polymer Reaction Design Group. Her research focuses on the design of facile polymer synthesis pathways, combining synthetic polymer chemistry, state-of-the-art polymer characterization and kinetic modelling. |
 Christopher Barner-Kowollik | Christopher Barner-Kowollik completed a PhD in Physical Chemistry at the University of Göttingen, before joining the Centre for Advanced Macromolecular Design at the University of New South Wales in Sydney, where he led a research team as Full Professor, after holding ranks from research associate to associate professor. He is currently Full Professor of Macromolecular Chemistry at the Karlsruhe Institute of Technology. His main research interests range from the synthesis of complex macromolecular architectures, their applications and material properties, the development of novel polymer and surface conjugation as well as polymerization protocols, quantitative mass spectrometry on polymer systems coupled with chromatographic techniques (multi-dimensional chromatography) to polymer reaction kinetics and mechanisms. |
1 Introduction
Nitrones, in addition to being known as 1,3-dipoles that undergo cycloaddition reactions,1 are known to be effective radical spin traps in electron spin resonance (ESR) experiments2 (see Scheme 1). Their dual nature has given rise to many useful applications not just in synthetic chemistry3 but also in biochemical systems as radical scavengers.4 Nonetheless, the majority of their applications has been limited to small molecule chemistry and only in recent years nitrones have been tried and applied in polymer chemistry, despite their established versatility. Interestingly, their ability to undergo cycloadditions is a frequent feature in synthetic organic chemistry while the potential to capture transient radicals appeared to be solely of interest to physical chemists. The combination of both to use the spin capturing ability as a tool for synthetic chemistry was until very recently only marginally addressed, which—given the high versatility, selectivity and tolerance to other functional groups of this reaction—comes as a surprise.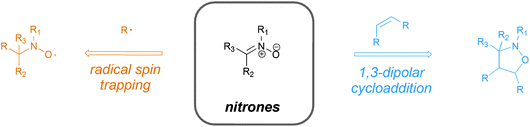 | ||
| Scheme 1 The versatility of nitrones in performing radical spin trapping and 1,3-dipolar cycloaddition reactions. | ||
The advent of controlled radical polymerization (CRP),5–9 which allows for the generation of polymeric materials with well-defined architectures and controllable properties, can be seen as a major contributing factor in expanding the use of nitrones towards macromolecular design. The fact that nitrones are efficient radical scavengers afforded them to be suitable candidates in mediating radical polymerizations to produce well-defined polymeric materials. Indeed, such a use as controlling agent became one of the earliest applications of nitrones in polymer chemistry.10,11 Concurrently, the development of nitronecycloaddition reactions on a macromolecular scale by Ritter and Vretik12 was equally significant towards this diversification process. Since then, a variety of techniques employing nitrones has continuously developed, such as the enhanced spin capturing polymerization (ESCP)13–18 and nitrone-mediated radical coupling (NMRC)19–21 reactions—all with the goal of providing variable avenues and novel platforms for advanced material synthesis.
The aim of the current review is to collate and summarize some of the past applications of nitrones in polymer synthesis as well as providing a state-of-the-art up-date focusing on the latest technologies and developments from our own laboratories, namely ESCP and NMRC. Comparisons between these two techniques with other selected methods reported in the literature with respect to their different synthetic applications will also be made herein. With the current review we hope to foster an awareness within the polymer community for the versatility of nitrones, whilst concomitantly demonstrating how nitrones can combine with existing technologies to produce tailor-made polymeric materials. As we will show, nitrone and nitroxide chemistries are closely related. It is, however, not the scope of this review to give an overview over the nitroxide mediated polymerization (NMP) technique, since NMP is mechanistically distinctly different from ESCP and NMRC. For an excellent review on NMP, the reader is referred to the literature.7
2 Early uses of nitrones in polymer synthesis
Some of the early pioneering applications of nitrones in polymer chemistry are mainly based on radical polymerization processes and as such, will be the main focus of this section. Other early applications will also be briefly discussed herein.2.1 Radical polymerization reactions
Since the late 1990s, several groups have independently reported on the utilization of nitrones in the context of radical polymerizations. These studies included the works by: (i) Grishin et al. who employed nitrones directly in reaction mixtures to control polymerization reactions at moderately low temperatures10 (e.g. 50 °C) and (ii) Jerome and co-workers, in whose work nitroxide radicals are formed in situ via the pre-reaction of nitrones with radical initiators at 85 °C before subsequent usage in NMP reactions at 110 °C.22In 1999, Braslau and Hawker et al.23 published work describing the synthesis of novel alkoxyamines that involved successive Grignard and catalytic oxidative reactions with nitrones to form nitroxides, followed by radical addition reactions. These alkoxyamines were purposefully designed such that a better level of control could be imparted on the polymerization of a more diverse range of monomers (besides styrene) such as acrylates and methacrylates, for which at that time no efficient control was achieved viaNMP. Later, similar approaches were adopted in synthesizing new alkoxyamines, also through synthetic modification of nitrone (and nitroso) compounds.24–26 Although the nitrones were only utilized as chemical precursors en route to synthesize alkoxyamines, these studies nonetheless demonstrated the indirect viability of using nitrones in polymer synthesis and perhaps provided the platform for other researchers to continue research in this field.
Grishin et al. polymerized methyl methacrylate (MMA) in the presence of a commercially available nitrone, N-tert-butyl-α-phenylnitrone (PBN), for which claims of an NMP reaction were made,10 even though the reaction temperature of 50 °C was well below the temperature required for the homolysis of the C–ON alkoxyamine bond. Regardless of the uncertainty of the reaction mechanism, these authors demonstrated that the polymerization reactions were better controlled compared to conventional free radical polymerizations in terms of the molecular weight. In addition, there was clear evidence of the suppression of the gel effect, a feature which is attractive for industrial processes.
In separate work, Jerome and co-workers initially described the pre-reaction of nitrones with radical initiators at 85 °C in the absence (and also tried in the presence) of monomers to form in situnitroxide/alkoxyamine compounds that are subsequently employed without further purification in the NMP of styrene at elevated temperatures (e.g. 110 °C). Additional studies27 were conducted by the same group employing an identical pre-reaction concept that led to the generation of well-defined copolymers, demonstrating the high efficiency of nitrones to impart the control on radical polymerization. Also, in one of these studies, the relevance of the pre-reaction procedure was highlighted.27d It was noted that the pre-reaction procedures of reacting nitrones with azoinitiators at 85 °C in the absence of monomers prior to NMP reactions (thus a two step process) afforded polymers with lower polydispersity index, PDI, values (1.25–1.35) compared to those (>1.65) obtained without this pre-reaction step. Judging solely from the PDI values, the method of pre-treating the nitrones was subsequently deemed to impart better control compared to techniques that directly utilise nitrones in polymerizable media. However, it should be stressed that higher PDI values do not necessarily imply that the process is poorly controlled, as one can still effectively control the molecular weight distributions of polymersvia nitrones without any pre-reaction steps. It is exactly such a concept that was later adopted by us and refined towards a control strategy for radical polymerizations which subsequently led to the enhanced spin capturing polymerization (ESCP) method that will be described below.
2.2 Other early polymer-based applications
During the 1990s, several researchers have developed similar techniques for generating polymeric materials with nitrone functionalities attached to the polymer backbone either for ESR spin trapping experiments28 or as photochemically active materials.29,30 The synthesis of these materials was generally accomplished by polymerizing aldehyde-based vinyl monomers via conventional free radical polymerization followed by condensation reactions of the poly(aldehyde) with excess N-alkyl hydroxylamines yielding poly(nitrones).28–30 These materials were generated for their specific aforementioned purposes at the time, although not much work was dedicated to such materials as follow-up work. Nevertheless, such poly(nitrones) may have the potential to spark applications in the synthesis field given the newer developments in macromolecular chemistries involving nitrones.Ritter and Vretik reported on a step growth mechanism via1,3-dipolar cycloaddition reactions between bifunctional nitrones and bis-maleimide compounds forming polymers that are inter-connected by isoxazolidine rings (see Scheme 2).12 No catalysts were used in these reactions, yet oxygen-free conditions and long reaction times (2 days) were required. These step growthpolymers were described to have good performance for coating formation on glass surfaces.12 The main feature of the above work is undoubtedly the successful cycloaddition reaction involving nitrones on a macromolecular scale. As the cycloadduct formed via this reaction often acts as a precursor to other useful functional groups such as β-lactams,31 the work of Ritter and co-workers certainly lays the foundation for new polymer functionalization techniques. Such cycloadditions are in fact no new chemistry. In the context of click chemistry32 and in view of the success of the analogue azide–alkynecycloaddition, nitrones as enophiles might become useful functionalities for polymerconjugation purposes. It is worth noting that an analogous cycloaddition reaction with nitrile oxides was recently developed as a tool for end grouppolymer modifications.33
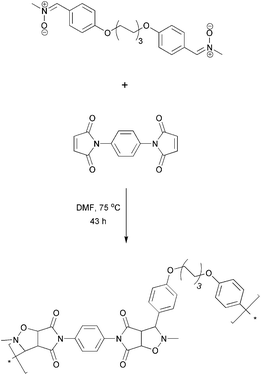 | ||
| Scheme 2 Step growthpolymerization of a bis-nitrone with a bis-maleimidevia a 1,3-dipolar cycloaddition reaction as reported by Vretik and Ritter.12 | ||
3 Enhanced spin capturing polymerization (ESCP)
Our interest in applying nitrones to mediate radical polymerizations arose from two earlier areas of interest: (i) the thioketone-mediated polymerization (TKMP)34 process, which employs thioketones as controlling agents via the formation of a reversible dormant radical species and (ii) the laser-induced marking of polymer chains in pulsed-laser polymerization systems.35 Based on these studies we have developed the so-called enhanced spin capturing polymerization (ESCP) method13–18 in which nitrones are used directly in the presence of a photo- or thermal initiator and monomer as a means of controlling radical polymerization processes. From an experimental point of view, the ESCP concept may seem identical to the aforementioned work by Grishin and co-workers. Despite the experimental similarities and to some extent even in the terms of the results that were reported by our colleagues,10 the underlying mechanism of ESCP is certainly not an NMP process. While alkoxyamines are formed during ESCP (see below for details), such formation is under normal reaction conditions irreversible and no equilibrium between an active and a dormant species is achieved or required to exert control over molecular weight or chemical functionality. Substantial dissociation of the alkoxyamine bond at temperatures below 80 °C (under which typical ESCP reactions are performed) is highly unlikely.36 We thus proposed a new mechanistic concept, which is supported by experimental data and kinetic studies.15 The details of the underlying mechanism of ESCP will be explained and discussed in the following sub-sections.3.1 Mechanism and kinetics of ESCP
The ESCP mechanism is based on the ability of the nitrones to limit the lifetime of a propagating radical and to enforce termination events by combination between propagating macroradicals via two sequential spin capturing reactions (see Scheme 3). The first spin capturing event is induced by the employed nitrone to form macronitroxide species which subsequently act as ‘secondary spin captors’ or ‘terminators’ towards a second propagating macroradical. The number of radicals in the system is thus reduced compared to the uncontrolled polymerization. In consequence, a lowered overall polymerization rate is observed, which is however, generally not untypical for many controlled polymerization reactions. As the reactions are routinely performed below 80 °C (or more precisely below the bond dissociation temperature of the generated alkoxyamine bond), the system resembles a controlled polymerization, but does not feature living characteristics (as for example in NMP) given the inability of the macronitroxide species to partake in any reversible reactions (i.e. the capping and uncapping actions between the nitroxide and transient radical species).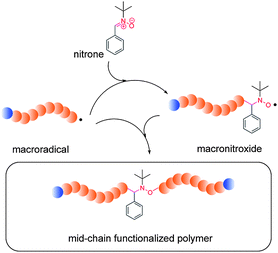 | ||
| Scheme 3 The enhanced spin capturing mechanism depicting the spin capturing of propagating macroradicals with N-tert-butyl-α-phenylnitrone, PBN, and the corresponding macronitroxide to form mid-chain functionalized polymers. Reproduced with permission from ref. 19. | ||
If the proposed mechanism of ESCP is operational, one would expect the alkoxyamine function to be situated in the middle of the polymer chains. In fact, this notion was proven via quenching experiments.15 Typically, in such an experiment, the polymer is dissolved in a suitable solvent (i.e.dimethylformamide) in the presence of a radical quencher such as tributyltin hydride. Subsequently, the reaction mixture is heated to 125 °C for 5 hours. Although the C–ON bond is not labile at lower temperatures, it is still inherently cleavable at higher temperatures (T > 100 °C). Thus—during the quenching procedure—the polymer chains literally break into half to form macroradicals and macronitroxides that are terminated by the excess amount of radical quenchers. An analysis of the products before and after the quenching sequence viasize exclusion chromatography (SEC) revealed the quenched samples having close to half the number average molecular weight of the original ESCP-generated polymer, thus indicating that the chains broke in the middle and that the alkoxyamine is truly incorporated into mid-chain position of an ESCP-derived polymer. This experimental result was also underpinned by kinetic modeling of the process on the basis of literature kinetic coefficients, by which the same result was derived.15 It is worth recognizing though that the ease in which the centered alkoxyamine can be cleaved at above 100 °C may render these polymers thermally unstable. On the other hand, certain alkoxyamine bonds that can exhibit higher thermal stability may limit its application such as the possibility of subsequent chain extensions viaNMP to form triblock copolymers (as described in a later section). Nevertheless, one can purposefully design the nitrone structure depending on the desired needs that either allow for post-NMP reactions or the formation of more stable alkoxyamines.
Despite the above proof of the presence of a mid-chain functional polymer—like in any other polymerization technique—there can be cases where the system may deviate from the ideal proposed mechanism. Thus, in order to maximise the formation of mid-chain functionalized polymersvia ESCP, a judicious choice of the radical initiator (or more precisely the attacking initiator radical fragment) is required. The selection of an appropriate attacking radical is critical due to the existence of competition reactions between the addition of small radical fragments from the initiator and the addition of propagating macroradicals to the nitrones to form either small nitroxides or macronitroxide species, respectively. To minimize the small nitroxide formation reaction that is caused by the initiator fragment, oxygen-centered radicals (e.g. derived from peroxideinitiatordecomposition) should be avoided and tertiary carbon-centered radicals (e.g. derived from azoinitiators) should be preferably used.15 This is simply due to the higher affinity of nitrones towards reacting with oxygen-centered radicals, followed by acrylates, styrenic and tertiary carbon-centered radicals.2a,13,17 Thus, when azoinitiators are employed in the polymerization of styrene or acrylate monomers, macronitroxides are formed instead of small nitroxides, thus leading to the formation of mid-chain functionalized polymers. Nevertheless, the initiator selection criterion is only important if a mid-chain functionalized polymer is required. An improper choice of the attacking radical (with respect to its ability to affect the formation of mid-chain functional alkoxyamines) does neither defeat the purpose of employing ESCP as a molecular weight control tool nor disprove its overall mechanism: a non-reversible enhanced (double) spin capturing reaction takes place regardless whether the radicals are originating from the initiator or propagating chains.
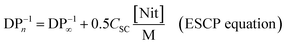
In terms of molecular weight control, ESCP bears a closer resemblance to classical chain transfer polymerization (CTP)37–39 than any other CRP technique. The control over molecular weight in ESCP is governed by the amount (i.e. concentration) of nitrone employed, which is also similar to CTP where the amount of chain transfer reagent (CTA) employed dictates the molecular weight of the generated polymers. Chain transfer reactions are the mediating cause in CTP, yet—from the perspective of the macroradicals—they are essentially terminated to form dead chains. Mechanistically, the CTP process is akin to the events occurring in ESCP, only that the propagating chains are terminated by the nitrones and the corresponding macronitroxides. Given that the fate of the macroradicals is similar in both systems, the kinetic equation derived for ESCP is—not surprisingly—almost identical to the Mayo equation. Similar to the Mayo equation employed to describe CTP, we have analogously defined a spin capturing constant, CSC, as the ratio of kad,macro/kp, where kad,macro and kp are the rate coefficients for the addition of macroradicals to the nitrones and propagation, respectively. Thus, by measuring the average molecular weight of polymers synthesized in the presence of various nitrone concentrations, the rate coefficient for the spin capturing of the macroradicals becomes accessible. The factor of 0.5 in the ESCP equation is included to take into account the fact that two macroradicals are terminated by one nitrone molecule. In fact, as the reaction product resembles a polymer generated by pure combination, the theoretical limit in dispersity is 1.5, which is also matched by experiment, while CTP generally yields polymers with a dispersity index around 2, which is typical for a distribution obtained from disproportionation. Thus, the spin capturing constant is the equivalent of the chain transfer constant in the Mayo equation.
The value of CSC has important implications for the ESCP process. For a CSC value close to unity (which leads to an equal consumption of nitrones and monomer during the course of the reaction), a constant Mn can be observed throughout the entire conversion regime. The same also applies to CTP in cases when the chain transfer constant, Ctr, is unity. Despite some of the apparent similarities noted above, it must be emphasized that both ESCP and CTP are still distinguishable from one another because: (i) the main mode of chain termination is intrinsically different (chain transfer in CTP and spin capturing in ESCP) and (ii) there are no re-initiating species present in ESCP as in CTP. Hence, ESCP polymers bear the initiator derived moieties as endgroups on the polymer whereas in CTP, the transferred functionality and the re-initiating fragment are primarily found.
One key factor which we have identified to be crucial in determining the CSC or kad,macro values is the nitrone structure.14 Investigations into the structure–reactivity correlations of different nitrones revealed that the type of N-alkyl groups or the attachment of different functionalities on the aromatic ring of phenylic-typed nitrones can have a significant effect on kad,macro. More bulky N-alkyl substituents such as N-tert-butyl result in lower reactivity compared to the less sterically hindered N-methylgroup. However, we have preferred to use the N-tert-butyl variants in most of our studies because of the better performance of such compounds in later NMP reactions in which methyl moieties would promote disproportionation and hence compromise the livingness of the reaction. Comparison of the determined kad,macro values for nitrones with various substituents on the aromatic structure clearly indicates that the attachment of electron-withdrawing groups enhances the nitrone reactivity, while electron donating groups impose the opposite, in agreement to what has been observed in independent studies.2a,27a It is of paramount importance to note that the simplest and most common method of synthesizing nitrones is via the condensation of aldehydes with N-alkyl hydroxylamines or via partial oxidation of secondary amines (see Scheme 4). The condensation reaction is in fact our most preferred choice of synthesis not just because of its simplicity and relatively high yield but also due to the ease in introducing chemical functionalities, for example, onto the aromatic ring of benzaldehyde educts, which allows for the facile synthesis of multifunctional nitrones.
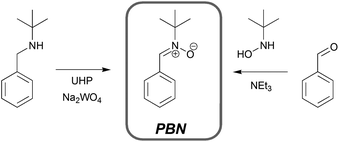 | ||
| Scheme 4 Synthesis pathways for the preparation of nitrones. | ||
3.2 Applications of ESCP
So far, it has been demonstrated how ESCP can be successfully employed for controlling polymerization reactions of styrene, butyl acrylate and N-isopropylacrylamide (NIPAAm) in either organic or aqueous solutions and via thermally or photo-chemically initiated chemical systems.13 In addition to these monomers, we have recently applied ESCP towards polymerizing methacrylate monomers albeit with the addition of 5–10 vol% of styrene comonomer.17 Obtaining good control over the polymerization of methacrylate monomers in nitroxide-related systems has often been challenging.23 One of the reasons is related to the propensity of the methacrylate radicals to disproportionate with the nitroxide species.40–42 There have been cases though where methacrylate monomers can be polymerized successfully viaNMP albeit via specially formed/designed nitroxide compounds.43–45 Another possible way of overcoming the difficulties in polymerizing this class of monomer is by adding small amounts of comonomer that would preferably favour combination reactions with nitroxide species, thus minimizing the event of disproportionations. Charleux and co-workers46 undertook such an approach successfully in NMP reactions. Inspired by their approach, a similar strategy was applied to ESCP which leads to successful mediation of MMA polymerization at 5% content of styrene as a comonomer, even though not so much the disproportionation reaction poses a problem in the ESCP of methacrylates rather than the lowered addition rate of the propagating chains onto the control agent.17 It should be noted that all above examples could be mediated by PBN, even polymerizations in water. The efficiency and versatility of ESCP in polymerizing different monomer systems under various reaction conditions could provide an attractive alternative to CTP, which is to date the industrially preferred method of molecular weight control.Other potential applications of ESCP include the possibility of utilizing the ESCP equation as a kinetic parameter estimation tool to determine the kad,macro of different monomer/nitrone pair systems (see Fig. 1). Such a methodology does not only complement current ESR techniques in determining addition rate coefficients of radicals to nitrones, yet also potentially allow for the determination of kad,macro for other monomer/radical spin traps systems (that obey the kinetics of ESCP), where ESR measurements are not feasible.
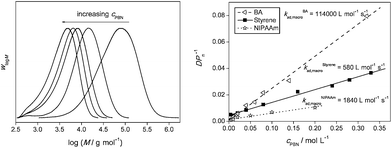 | ||
| Fig. 1 Molecular weight distributions obtained for ESCP of polystyrene at different nitrone concentrations (left) and determination of kad,macro for different monomer/nitrone pair systems utilizing the ESCP equation (right). Reproduced with permission from ref. 13 and 14. | ||
4 Nitrone-mediated radical coupling (NMRC) reactions
In recent years, the employment of efficient polymer–polymerconjugation chemistries47–49 as macromolecular engineering tools has received great interest among polymer scientists. As a result, significant effort has been dedicated towards developing novel conjugation chemistries, amongst which are also radical-based coupling reactions.50–52 This includes the newly developed nitrone-mediated radical coupling (NMRC)19 reaction, which will be described in the following.The main mechanism of NMRC is exactly identical to the spin capturing concept of ESCP, i.e. the radical coupling of two polymeric strands with active radical centers via the nitrone (refer to Scheme 3). However, the difference between the two techniques lies in the source of the macroradicals. In ESCP, macroradicals are generated during the course of the radical polymerization, whereas in NMRC macroradicals are stored in the form of living polymers already pre-made via a living free radical polymerization technique (e.g. atom transfer radical polymerization (ATRP)). NMRC thus yields polymers that are structurally identical to the ESCP product, with the added advantage of obtaining narrow molecular weight distributions.
Two model polymer systems, polystyrene and poly(isobornyl acrylate) prepared viaATRP, have so far been tested for NMRC reactions yielding highly functionalized (>90%) polymers with precisely centered-alkoxyamine functions. The extent of coupling is influenced by two main factors: (i) the rate of activation/deactivation for releasing the macroradicals and (ii) the kad,macro value for the type of monomer/nitrone pair system. The kad,macro values for styrenic-based macroradicals are two orders of magnitude lower than those for acrylate-type radicals. Thus, to obtain high coupling efficiencies for styrenics, slightly larger nitrone concentrations (up to 5 eq.) are required or a slower release of macroradicals via a less active catalytic system, respectively, whereas acrylate-type polymers can be coupled with equimolar amounts of the nitrone.
The development of NMRC complements some of the other radical–radical coupling reactions that are available in the literature, such as the atom transfer radical coupling (ATRC),50cobalt mediated radical coupling (CMRC)51 and nitroxide radical coupling (NRC).52NMRC, ATRC and CMRC have in common—in terms of their general mechanism—that two polymer chains are activated and coupled in situ via combination events to form homotelechelic polymers with specific mid-chain functionalities. However, it must be emphasized that the individual coupling chemistries are different with respect to each method (i.e.nitrone spin capturing in NMRC; conventional bimolecular termination in ATRC; and diene-assisted radical coupling in CMRC). It should be noted that NMRC has a distinct advantage over ATRC, that is, termination in NMRC only occurs via combination reaction. Hence, also systems that undergo termination via disproportionation can be coupled whereas the same target cannot be reached with ATRC in such cases. Despite the efficiency of all different coupling reactions, it remains a synthetic challenge if one were to couple two distinctively different polymer precursors of different compositions in a controlled fashion via any of these three methods. With NRC reactions, the nitroxide functionality is incorporated at the end of a specific-type of polymer (or multifunctional compound) and can react with other types of carbon-centered macroradicals besides its own kind. Thus, from a synthetic point of view, the advantages of an NRC reaction are that it may allow for a more favourable synthetic approach towards copolymers and heterotelechelic structures. This synthetic advantage is, however, bought in via an additional synthesis step, e.g. the synthesis of a macronitroxide species, which is not always easily available.
5 Complex macromolecular architectures via ESCP or NMRC
The following section will detail the different types of macromolecular architectures that have been prepared via ESCP or NMRC to date. These are ABA-type block copolymers; star polymers; ‘step growth’ polymers; and dendritic macromolecules.5.1 Block copolymers
Although the proposed ESCP and even NMRC reactions do not fall into the category of a living system due to the absence of equilibrium reactions between the nitroxide and free radical species under the employed reaction conditions, the generated mid-chain functionalized polymers are essentially macroalkoxyamines and can behave as macroinitiators in subsequent NMP reactions (at temperatures > 100 °C). Therefore, purified ESCP- and NMRC-derived polymers can re-initiate the polymerization with other monomers under NMP conditions to produce ABA′-type triblock copolymers (see Fig. 2).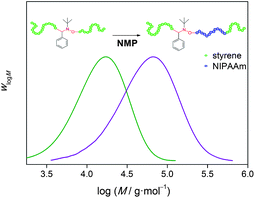 | ||
| Fig. 2 SEC distributions elucidating a successful sequential ESCP–NMP reaction to produce ABA′-type triblock copolymers. Reproduced with permission from ref. 15. | ||
We demonstrated that polystyrene prepared via ESCP allowed the subsequent NMP of butyl acrylate and NIPAAm to produce poly(styrene)-b-poly(butyl acrylate)-b-poly(styrene) and poly(styrene)-b-poly(NIPAAm)-b-poly(styrene) copolymers accordingly.13,15 In addition, we have also monitored the kinetics in the NMP of styrenevia on-line FT-NIRspectroscopy and off-line SEC, showing a linear increase in Mn with conversion indicating good living characteristics, provided the reaction conditions are chosen accordingly.18
NMRC products may also be directly employed to produce triblock copolymers, even though no experimental study has been carried out on these systems so far. The reactivity of the embedded alkoxyamine functionalities will, however, be largely independent of the broadness of the individual ESCP or NMRC distribution. Alternatively, narrow ABA-type structures can be achieved if a diblock copolymer precursor, for instance (i.e. prepared via two consecutive ATRP reactions to form an AB copolymer with a halide termini), is subjected to nitrone spin capturing reactions to generate a final polymer product with A′B′BA composition. This approach—although also not yet undertaken—is in line with work that has been published by Detrembleur and co-workers where poly(vinyl acetate)-b-poly(acrylonitrile) polymers preformed by cobalt-mediated radical polymerization were subjected to CMRC reactions to produce triblock copolymers.51 Thus, even the production of A′B′CBA-type pentablock copolymersvia tandem NMRC–NMP may become possible in the future.
5.2 Miktoarmed stars
The general trend in the synthesis of complex star block copolymers53 nowadays is often aided by the combination of various efficient polymer–polymer ligation chemistries that can be classified as click chemistry.32The synthesis of miktoarmed star polymersvia ESCP or NMRC was achieved through a specially designed nitrone that contains a secondary alkyne functionality allowing for post-click reaction with polymers bearing azide end groups.16Polystyrene and poly(isobornyl acrylate) prepared via ESCP and NMRC respectively with this designated nitrone have been successfully modified by copper-catalyzed azide–alkynecycloaddition (CuAAC) reactions with azide-terminated polystyrene and/or poly(isobornyl acrylate) to generate 3-armed star (co)polymers (see Scheme 5) both via ESCP and NMRC, respectively.
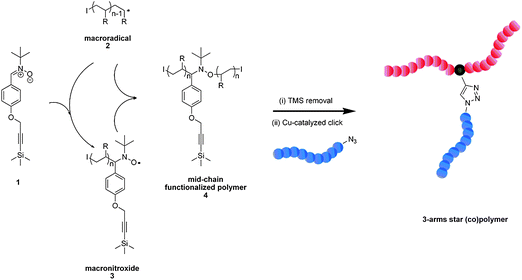 | ||
| Scheme 5 Schematic illustrating the tandem ESCP/NMRC–CuAAC strategy in generating miktoarmed star polymers. (I denotes the polymer end group resulting from the employed initiator and R represents the monomer side group—COOR for isobornyl acrylate or phenyl for styrene). Reproduced with permission from ref. 16. | ||
With reference to the above work, it was possible to demonstrate that the incorporation of click functions onto nitrones is feasible in a facile manner and that the control agent can be used as a carrier to form polymers with a specific mid-chain functionality, as both ESCP and NMRC are tolerable towards other functionalities. Thus, in essence, the synthesis of complex architectures via a combination of ESCP/NMRC and click chemistry can potentially be expanded beyond the realm of star polymers. The limitation of the above possibility would in fact be more likely depend on the effectiveness and the flexibility of the click reaction itself rather than on ESCP/NMRC. However, such limitations also apply to other synthetic strategies that rely on the technology of click chemistry.
5.3 Polymers with embedded functionalities
One of the latest developments in our laboratories is the utilization of NMRC in combination with α,ω-bromo functionalized poly(isobornyl acrylate) in a novel step growthpolymerization (see Scheme 6).20 The model polymer precursor was prepared viaATRP of isobornyl acrylate with dimethyl 2,6-dibromoheptanedioate as a bifunctional initiator. NMRC reaction of this polymer yielded ‘step growth’ polymers with an average of only 5–6 ‘monomer’ units and interesting synthetic features, i.e. the embodiment of alkoxyamine functions at specific sites along the polymer backbone. Thus, the distance between each functionality in the polymer chain can be tuned precisely by simply using different polymer precursors pre-made with the desired chain lengths.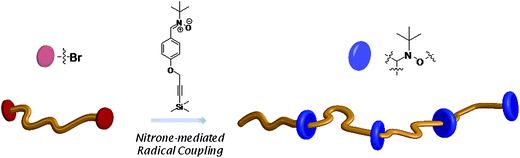 | ||
| Scheme 6 Generation of polymers with site-specific functionalities viaNMRC of α,ω-bromo-terminated polymers with nitrones. Reproduced with permission from ref. 20. | ||
Besides creating polymer chains with periodic incorporation of alkoxyamine functions, NMRC of bifunctional polymers with ‘clickable’ nitrones also allows for the insertion of secondary site-specific functionalities onto the polymer chain. The same model dibromo poly(isobornyl acrylate) (see above) was subjected to NMRC reactions with an alkyne-functionalized nitrone to introduce alkyne functions on the backbone of the coupled product. As a proof of concept, successful model post-modifications of the alkyne were demonstrated viathiol–yne reactions with 3-mercaptopropionic acid. This latest application viaNMRC exemplifies once more the efficiency of the underlying spin capturing reactions and compliments other methods that are available and also employ a step growth approach advantageously to produce site-specific functionalized polymers.54–56
5.4 Dendrimers
In another very recent approach, we have developed a novel dendrimer synthesis methodology based on a combination of NMRC reactions and classical click chemistry (Scheme 7).21 The capability of each nitrone to react with two radical centers allows to build an ordered higher generation structure as a result of the doubling of functional groups in its reaction. Thereby the newly generated mid-chain functionalized alkoxyamine serves as the branching point.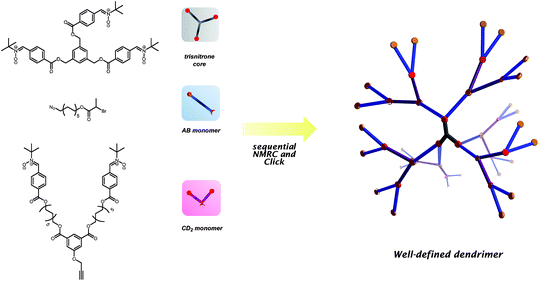 | ||
| Scheme 7 Illustration of the general concept in combining NMRC with click chemistry to produce well-defined dendrimers. Reproduced with permission from ref. 21. | ||
A trisnitrone core was synthesized and reacted with an azide-functionalized ATRPinitiator (AB-monomer) via NMRC to generate first generation dendrimers, G1-[N3]6. A CD2-typed monomer that contains two nitrones at both ends with a centered alkyne functionality was synthesized and clicked with G1-[N3]6viaCuAAC yielding second generation dendrimersG2-[Nit]12. A second NMRC reaction was applied with the AB-monomer to produce azide-terminated dendrimers, G3-[N3]24 that can be further functionalized viaCuAAC when desired to generate tailor-made dendritic structures.
During the above synthesis, only minimal column purifications were required underpinning the efficiency of the reactions. In addition, each modification step (NMRC or click) resulted in an increase in generation and can thus be considered as an accelerated-synthesis—placing this method on par with other efficient dendrimer synthesis chemistries such as thiol–ene57 and thiol–yne.58 Thus, this application further demonstrates the versatility of nitrones in synthetic (macromolecular) chemistry.
6 Conclusions and outlook
The increase in the number of applications of nitrones in the field of macromolecular chemistry over the last few years is indicative of the promising potential of these effective yet—in polymer chemistry—lesser acknowledged compounds. The emergence of other technologies, most notably CRP and click chemistry—intended to meet the high demand for advanced synthetic materials—also played a crucial role in this expansion process. Nonetheless, the examples presented herein merely constitute a fraction of the types of chemistry that can be carried out with nitrones, as there are many other branches of chemistry (e.g. surface chemistry, photochemistry, bioconjugated polymeric nitrones, etc.) that are still waiting to be explored. Due to their efficiency in spin capturing, many options for the design of new synthesis avenues open up, which are not restricted to polymer chemistry as the synthesis of dendrimers demonstrated.It goes without saying that only through innovation and creativity new and more advanced synthetic avenues can be made possible, as evidenced by the technological development of nitrones. We do, however, not consider nitronepolymer chemistry to be any superior or inferior to other available synthetic chemistries, but rather as a part of a broader family of molecular engineering tools that can contribute to the advancement of modern polymer science and technology. We hope that the current review serves as a stimulus to our colleagues to explore the use of nitrones in their own synthetic endeavours.
Abbreviations
| ATRC | atom transfer radical coupling |
| ATRP | atom transfer radical polymerization |
| BA | butyl acrylate |
| CMRC | cobalt-mediated radical coupling |
| CRP | controlled radical polymerization |
| C SC | spin capturing constant |
| CTA | chain transfer agent |
| CTP | chain transfer polymerization |
| C tr | chain transfer constant |
| CuAAC | copper-catalyzed alkyne–azidecycloaddition |
| DMF | dimethylformamide |
| DP | degree of polymerization |
| ESCP | enhanced spin capturing polymerization |
| ESR | electron spin resonance |
| FT-NIR | Fourier transform-near infrared |
| Gn | Dendrimers with nth generation |
| k ad,macro | rate coefficient of the addition of macroradicals to nitrone |
| k p | propagation rate coefficient |
| M | vinyl monomer (general) |
| MMA | methyl methacrylate |
| M n | average molecular weight |
| N3 | azide |
| NIPAAm | N-isopropylacrylamide |
| Nit | nitrone |
| NMRC | nitrone-mediated radical coupling |
| NMP | nitroxide-mediated polymerization |
| NRC | nitroxide radical coupling |
| PBN | N-tert-butyl-α-phenylbutylnitrone |
| PDI | polydispersity index |
| SEC | size exclusion chromatography |
| Sty | styrene |
| TKMP | thioketone-mediated polymerization |
| UHP | urea hydrogen peroxide |
References
- (a) See for example: K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863–909 Search PubMed; (b) J. Hamer and A. Macaluso, Chem. Rev., 1964, 64, 473–495 CrossRef CAS; (c) D. S. C. Black, R. F. Crozier and V. C. Davis, Synthesis, 1975, 205–221 CrossRef CAS.
- (a) See for example: T. J. Kemp, Prog. React. Kinet. Mech., 1999, 24, 287–358 Search PubMed; (b) E. G. Janzen, P. H. Krygsman, D. A. Lindsay and D. L. Haire, J. Am. Chem. Soc., 1990, 112, 8279–8284 CrossRef CAS; (c) E. Finkelstein, G. M. Rosen and E. J. Rauckman, J. Am. Chem. Soc., 1980, 102, 4994–4999 CrossRef CAS; (d) E. G. Janzen, Y. Y. Wang and R. V. Shetty, J. Am. Chem. Soc., 1978, 100, 2923–2925 CrossRef CAS; (e) E. G. Janzen, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS.
- (a) See for example: G. Ghini, L. Luconi, A. Rossin, C. Bianchini, G. Giambastiani, S. Cicchi, L. Lascialfari, A. Brandi and A. Giannasi, Chem. Commun., 2010, 46, 252–254 Search PubMed; (b) C. S. Mckay, J. Moran and J. P. Pezacki, Chem. Commun., 2010, 46, 931–933 RSC; (c) J. J. Tufariello, Acc. Chem. Res., 1979, 12, 396–403 CrossRef CAS; (d) R. C. F. Jones and J. N. Martin, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, Wiley & Sons, New York, 2002 Search PubMed; (e) I. A. Grigor'ev, in Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis, ed. H. Feuer, Wiley & Sons, Hoboken, 2008 Search PubMed; (f) A. Dondoni, S. Franco, F. Junquera, F. L. Merchan, P. Merino and T. Tejero, J. Org. Chem., 1997, 62, 5497–5507 CrossRef CAS; (g) K. B. Chakraborty, G. Scott and H. Yaghmour, J. Appl. Polym. Sci., 1985, 30, 3267–3281 CrossRef CAS.
- (a) See for example: R. A. Floyd, R. D. Kopke, C.-H. Choi, S. B. Foster, S. Doblas and R. A. Towner, Free Radical Biol. Med., 2008, 45, 1361–1374 Search PubMed; (b) R. A. Floyd, Free Radical Biol. Med., 2009, 46, 1004–1013 CrossRef CAS; (c) C. E. Thomas, D. F. Ohlweiler, V. L. Taylor and C. J. Schmidt, J. Neurochem., 1997, 68, 1173–1182 CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- G. Moad and C. Barner-Kowollik, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008 Search PubMed.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- D. F. Grishin, L. L. Semenycheva and E. V. Kolyakina, Russ. J. Appl. Chem., 2001, 74, 483–486 CrossRef.
- M. V. Pavlovskaya, E. V. Kolyakina, V. V. Polyanskova, L. L. Semenycheva and D. F. Grishin, Russ. J. Appl. Chem., 2002, 75, 1905–1909.
- L. Vretik and H. Ritter, Macromolecules, 2003, 36, 6340–6345 CrossRef CAS.
- E. H. H. Wong, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7273–7279 CrossRef CAS.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1098–1107 CrossRef CAS.
- T. Junkers, E. H. H. Wong, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2009, 42, 5027–5035 CrossRef CAS.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 3785–3793 CrossRef CAS.
- L. Zang, E. H. H. Wong, C. Barner-Kowollik and T. Junkers, Polymer, 2010, 51, 3821–3825 CrossRef CAS.
- L. Zang, E. H. H. Wong, C. Barner-Kowollik and T. Junkers, preparation.
- E. H. H. Wong, C. Boyer, M. H. Stenzel, C. Barner-Kowollik and T. Junkers, Chem. Commun., 2010, 46, 1959–1961 RSC.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., submitted Search PubMed.
- E. H. H. Wong, O. Altintas, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, Chem. Commun., submitted Search PubMed.
- C. Detrembleur, V. Sciannamea, C. Koulic, M. Claes, M. Hoebeke and R. Jerome, Macromolecules, 2002, 35, 7214–7223 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- M.-O. Zink, A. Kramer and P. Nesvadba, Macromolecules, 2000, 33, 8106–8108 CrossRef CAS.
- R. B. Grubbs, J. K. Wegrzyn and Q. Xia, Chem. Commun., 2005, 80–82 RSC.
- A. Studer, K. Harms, C. Knoop, C. Muller and T. Schulte, Macromolecules, 2004, 37, 27–34 CrossRef CAS.
- (a) V. Sciannamea, A. Guerrero-Sanchez, U. S. Schubert, J.-M. Catala, R. Jerome and C. Detrembleur, Polymer, 2005, 46, 9632–9641 CrossRef CAS; (b) V. Sciannamea, M. Bernard, J.-M. Catala, R. Jerome and C. Detrembleur, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7273–7279 CrossRef CAS; (c) V. Sciannamea, J.-M. Catala, R. Jerome and C. Detrembleur, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1219–1235 CrossRef CAS; (d) V. Sciannamea, J.-M. Catala, R. Jerome, C. Jerome and C. Detrembleur, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1085–1097 CrossRef CAS; (e) V. Sciannamea, R. Jerome and C. Detrembleur, Chem. Rev., 2008, 108, 1104–1126 CrossRef CAS.
- J. Ren, K. Sakakibara and M. Hirota, React. Polym., 1994, 22, 107–114 CrossRef CAS.
- K. Tanaka, T. Igarashi and T. Sakurai, Macromolecules, 2004, 37, 5482–5484 CrossRef CAS.
- M. Heinenberg and H. Ritter, Macromol. Chem. Phys., 1999, 200, 1792–1805 CrossRef CAS.
- (a) See for example: A. Padwa, K. F. Koehler and A. Rodriguez, J. Am. Chem. Soc., 1981, 103, 4974–4975 Search PubMed; (b) A. Pernet-Poil-Chevrier, F. Cantagrel, K. L. Jeune, C. Philouze and P. Y. Chavant, Tetrahedron: Asymmetry, 2006, 17, 1969–1974 CrossRef CAS; (c) T. Tejero, A. Dondoni, I. Rojo, F. L. Merchan and P. Merino, Tetrahedron, 1997, 53, 3301–3318 CrossRef CAS.
- (a) See for example: H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 Search PubMed; (b) C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987–992 CrossRef CAS; (c) J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS; (d) U. Mansfeld, C. Pietsch, R. Hoogenboom, C. Remzi Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560–1598 RSC; (e) C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS.
- (a) I. Singh, Z. Zarafshani, F. Heaney and J.-F. Lutz, Polym. Chem., 2011 10.1039/c0py00236d; (b) I. Singh, Z. Zarafshani, J.-F. Lutz and F. Heaney, Macromolecules, 2009, 42, 7709–7717 CrossRef CAS.
- (a) A. Ah Toy, H. Chaffey-Millar, T. P. Davis, M. H. Stenzel, E. I. Izgorodina, M. L. Coote and C. Barner-Kowollik, Chem. Commun., 2006, 835–837 RSC; (b) H. Chaffey-Millar, E. I. Izgorodina, C. Barner-Kowollik and M. L. Coote, J. Chem. Theory Comput., 2006, 2, 1632–1645 CrossRef CAS; (c) T. Junkers, M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 746–753 CrossRef CAS; (d) F. Gunzler, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1864–1876.
- T. Junkers, E. H. H. Wong, Z. Szablan, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2008, 29, 503–510 CrossRef CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS.
- J. Chiefari and E. Rizzardo, in Handbook of Radical Polymerization, ed. K. Matyjaszewski and T. P. Davis, Wiley-Interscience, Hoboken, 2002 Search PubMed.
- H. R. Snyder, J. M. Stewart, R. E. Allen and R. J. Dearborn, J. Am. Chem. Soc., 1946, 68, 1422–1428 CrossRef.
- (a) F. R. Mayo, J. Am. Chem. Soc., 1943, 65, 2324–2329 CrossRef CAS; (b) R. A. Gregg and F. R. Mayo, J. Am. Chem. Soc., 1948, 70, 2373–2378 CrossRef CAS.
- G. S. Ananchenko and H. Fischer, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3604–3621 CrossRef CAS.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Tordo and D. Bertin, Macromol. Chem. Phys., 2006, 207, 1278–1288 CrossRef CAS.
- C. Dire, J. Belleney, J. Nicolas, D. Bertin, S. Magnet and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6333–6345 CrossRef CAS.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Astolfi, L. Greci, P. Tordo and D. Bertin, Macromolecules, 2007, 40, 3108–3114 CrossRef CAS.
- C. Detrembleur, Ph. Teyssie and R. Jerome, Macromolecules, 2002, 35, 1611–1621 CrossRef CAS.
- A. C. Greene and R. B. Grubbs, Macromolecules, 2009, 42, 4388–4390 CrossRef CAS.
- (a) J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34–47 CrossRef CAS; (b) J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274–8282 CrossRef CAS; (c) J. Nicolas, L. Mueller, C. Dire, K. Matyjaszewski and B. Charleux, Macromolecules, 2009, 42, 4470–4478 CrossRef CAS.
- (a) A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CAS; (b) S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC; (c) A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1792–1798 CrossRef CAS.
- (a) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS; (b) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- R. A. Evans, Aust. J. Chem., 2007, 60, 384–395 CrossRef CAS.
- (a) B. Otazaghine, C. Boyer, J.-J. Robin and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2377–2394 CrossRef CAS; (b) T. Sarbu, K.-Y. Lim, J. Ell, D. J. Siegwart, J. Spanswick and K. Matyjaszewski, Macromolecules, 2004, 37, 3120–3127 CrossRef CAS; (c) T. Sarbu, K.-Y. Lim, J. Spanswick, R. R. Gil, D. J. Siegwart and K. Matyjaszewski, Macromolecules, 2004, 37, 9694–9700 CrossRef CAS.
- (a) A. Debuigne, C. Jerome and C. Detrembleur, Angew. Chem., Int. Ed., 2009, 48, 1422–1424 CrossRef CAS; (b) A. Debuigne, R. Poli, J. De Winter, P. Laurent, P. Gerbaux, P. Dubois, J.-P. Wathelet, C. Jerome and C. Detrembleur, Chem.–Eur. J., 2010, 16, 1799–1811 CrossRef CAS; (c) A. Debuigne, R. Poli, J. De Winter, P. Laurent, P. Gerbaux, J.-P. Wathelet, C. Jerome and C. Detrembleur, Macromolecules, 2010, 43, 2801–2813 CrossRef CAS.
- (a) Q. Fu, Z. Zhang, W. Lin and J. Huang, Macromolecules, 2009, 42, 4381–4383 CrossRef CAS; (b) C. Liu, M. Pan, Y. Zhang and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6754–6761 CrossRef CAS; (c) W. Lin, Q. Fu, Y. Zhang and J. Huang, Macromolecules, 2008, 41, 4127–4135 CrossRef CAS; (d) Q. Fu, G. Wang, W. Lin and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 986–990 CrossRef CAS; (e) J. Kulis, C. A. Bell, A. S. Micallef, Z. Jia and M. J. Monteiro, Macromolecules, 2009, 42, 8218–8227 CrossRef CAS.
- (a) See for example: N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 857–871 Search PubMed; (b) P. Moschogianni, S. Pispas and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 650–655 CrossRef CAS; (c) O. Altintas, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5916–5928 CrossRef CAS; (d) O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3588–3598 CrossRef CAS; (e) O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5699–5707 CrossRef CAS; (f) O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1218–1228 CrossRef CAS.
- Y.-G. Lee, Y. Koyama, M. Yonekawa and T. Takata, Macromolecules, 2009, 42, 7709–7717 CrossRef CAS.
- F. A. Plamper, S. Reinicke, M. Elomaa, H. Schmalz and H. Tenhu, Macromolecules, 2010, 43, 2190–2203 CrossRef CAS.
- (a) K. Satoh, M. Matsuda, K. Nagai and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 10003–10005 CrossRef CAS; (b) M. Mizutani, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 7498–7507 CrossRef CAS; (c) K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120–5156 CrossRef CAS.
- (a) K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS; (b) T. Kang, R. J. Amir, A. Khan, K. Ohshimizu, J. N. Hunt, K. Sivanandan, M. I. Montanez, M. Malkoch, M. Ueda and C. J. Hawker, Chem. Commun., 2010, 46, 1556–1558 RSC; (c) P. Antoni, M. J. Robb, L. Campos, M. Montanez, A. Hult, E. Malmstrom, M. Malkoch and C. J. Hawker, Macromolecules, 2010, 43, 6625–6631 CrossRef CAS.
- G. Chen, J. Kumar, A. Gregory and M. H. Stenzel, Chem. Commun., 2009, 6291–6293 RSC.
| This journal is © The Royal Society of Chemistry 2011 |