Construction of mixed micelle with cross-linked core and dual responsive shells†
Cong
Chang‡
,
Hua
Wei‡
,
Qian
Li
,
Bin
Yang
,
Ni
Chen
,
Jin-Ping
Zhou
,
Xian-Zheng
Zhang
* and
Ren-Xi
Zhuo
Key Laboratory of Biomedical Polymers of Ministry of Education & Department of Chemistry, Wuhan University, Wuhan, 430072, P. R. China. E-mail: xz-zhang@whu.edu.cn; Fax: +86-27-68754509; Tel: +86-27-68754509
First published on 12th January 2011
Abstract
A core cross-linked (CCL) mixed micelle with dual responsive shells was constructed from two amphiphilic block copolymers poly(methyl methacrylate-co-3-(trimethoxysilyl)propyl methacrylate)-b- poly(N-isopropylacrylamide) (P(MMA-co-MPMA)-b-PNIPAAm) and P(MMA-co-MPMA)-b-poly(2-(diethylamino)ethyl methacrylate) (P(MMA-co-MPMA)-b-PDEA) via a two-step process: cooperative aggregation of the two block copolymers into core-shell mixed micelles in acidic aqueous solution at room temperature followed by cross-linking of the hydrophobic core via an acid-catalyzed sol–gel process. The reversibly structural transformation of the core-shell mixed micelles into core-shell-corona (CSC) mixed micelles took place when subjected to elevated temperature or pH value, that is, high temperature resulted in the fabrication of CSC mixed micelle with shrunk PNIPAAm chains as the inner shell and stretched PDEA chains as the outer corona, and alkaline pH led to the formation of CSC mixed micelle with collapsed PDEA chains as the inner shell and extended PNIPAAm chains as the outer corona. Due to the existence of thermo- and pH- dually responsive shells, the structurally stable CCL mixed micelle may find practical applications in biomedical fields such as drug delivery and intelligent release.
Introduction
During the past decade, great interest has been focused on various materials suitable for biomedical applications, especially about the smart polymeric micelles due to their unique core-shell structures and stimulus responsive properties.1–5 Up to now, numerous thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) (PNIPAAm) have been reported,6 and their thermo-sensitivity has been frequently combined with pH-sensitivity for the purpose of developing multifunctional micelles exhibiting double stimulus-responsivities.7–9 As far as we know, double responsive micelles are usually prepared from dual sensitive random or block copolymers consisting of thermo-responsive moiety and pH sensitive segment simultaneously.10–13 Compared to this conventional strategy, another effective alternative leading to dual responsive micelles is the fabrication of mixed micelles by using cooperative self-assembly of two different block copolymers in their common selective solvent. Construction of mixed micelles possessing multicomponents is a more promising and facile approach to produce multifunctional micelles due to the intriguing co-micellization behaviors, therefore has attracted a great deal of recent attention. Moreover, the structure and property of mixed micelles can be conveniently tailored by varying the relative content of the two block copolymers in the mixed composition.14–16 The A–B and A–C diblock copolymers, which have a common hydrophobic block A and different hydrophilic blocks B and C, are able to co-self-assemble into mixed micelles in an aqueous medium.17,18The atom transfer radical polymerization (ATRP) technique, as one of the most actively developing areas in polymer chemistry, allows the facile preparation of a broad range of polymers with well-controlled molecular weight and preselected composition, especially amphiphilic block copolymers with well-defined structure and functional groups, without the stringent requirement which is usually necessary for other types of living polymerization.19–22
On the basis of these concepts, herein we report the synthesis of two well-defined diblock copolymers: temperature-responsive poly(methyl methacrylate-co-3-(trimethoxysilyl) propylmethacrylate)-b-poly(N-isopropylacrylamide) (P(MMA-co-MPMA)-b-PNIPAAm) and pH-responsive P(MMA-co-MPMA)-b-poly(2-(diethylamino)ethyl methacrylate) (P(MMA-co-MPMA)-b-PDEA) with the same hydrophobic moiety of PMMA via successive ATRP, and further proposed a novel mixed micelle structure with dual responsive outer shells self-assembled from these two amphiphilic stimulus-responsive block copolymers. The resultant mixed micelle was composed of a hydrophobic PMMA core and combined temperature/pH dual responsive PNIPAAm/PDEA shells (Fig. 1).
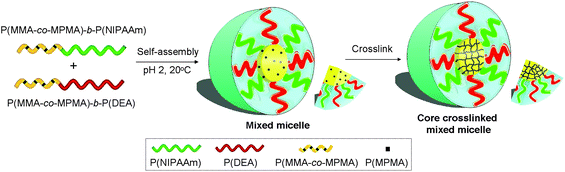 | ||
| Fig. 1 Schematic illustration of the CCL mixed micelle formation. Coaggregation of P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA in aqueous solution followed by cross-linking of the micellar core. | ||
In addition, the structural stability and integrity of the supramolecular nanostructures is a crucial issue in the practical applications of current micelle formulation. In this work, MPMA bearing reactive trimethoxysilyl functions was introduced to the PMMA block as a comonomer to produce the core cross-linked (CCL) mixed micelle by using inorganic “silica-based” cross-linking strategy,23–25 which can fix the structure of the mixed micelle and permanently suppress the dissociation of non-cross-linked micelle into individual polymer chains under shear forces, dilution by gastric, blood or other body fluids and salinity fluctuations after administration.26–28
By increasing temperature or pH value, PNIPAAm and PDEA blocks were respectively rendered hydrophobic, therefore the resulting core-shell mixed micelle converted into core-shell-corona (CSC) mixed micelle with three-layered structure, namely, the shrunk PNIPAAm chains formed the inner shell and the stretched PDEA chains constructed the outer corona at elevated temperature, and the collapsed PDEA chains built the inner shell and the extended PNIPAAm chains constituted the outer corona at alkaline pH. This study, to our knowledge, is believed to present the first example of CCL mixed micelle with dual responsive shells.
Experimental section
Materials
N-isopropylacrylamide (NIPAAm), ethyl 2-bromoisobutyrate (EBB) and tris(aminoethyl)amine (TREN) were purchased from Acros and used as received. Tris(2-(dimethylamino)ethyl)amine (Me6TREN) was synthesized from TREN according to previous study by Ciampolini and Nardi.292-(Diethylamino)ethyl methacrylate (DEA, Aldrich) was vacuum-distilled prior to use. 3-(Trimethoxysilyl)propyl methacrylate (MPMA) was purchased from Wuhan University Chemical Plant (Wuhan, China) and used as supplied. Methyl methacrylate (MMA) and N,N′-dimethylformamide (DMF), purchased from Shanghai Chemical Reagent Co. (Shanghai, China), were used after distillation under reduced pressure. CuBr (Shanghai Chemical Reagent Co.) was purified by stirring in acetic acid, washing with acetone, and then drying under vacuum. 2,2′-bipyridine (bpy), n-hexane, ethyl ether and all other chemicals were purchased from Shanghai Chemical Reagent Co. and used without further purification.Preparation of P(MMA-co-MPMA)-Br macroinitiator
The macroinitiator P(MMA-co-MPMA)-Br was prepared by ATRP of MMA and MPMA using EBB as the initiator and CuBr/bpy as the catalyst. A typical synthesis procedure was introduced as follows. A reaction flask with a magnetic stirrer was charged with EBB (146.8 μL, 1 mmol), MMA (5.336 mL, 50 mmol), MPMA (238.8 μL, 1 mmol), bpy (312.38 mg, 2 mmol), and DMF (5 mL). The flask was degassed by three cycles of freezing–pumping–thawing, back-filled with N2, and then CuBr (143.45 mg, 1 mmol) was introduced into the flask under the protection of N2 flow. After another three freeze–pump–thaw cycles, the reaction mixture was sealed followed by immersing the flask into an oil bath preheated at 70 °C to start the polymerization. After polymerization for 15 h, the mixture was diluted with 5 mL of DMF and the copper catalyst was removed by filtering the solution through a short Al2O3 column. The resulting solution was concentrated and poured into n-hexane to precipitate the product. The product was separated by filtration and further purified by redissolving/reprecipitating with tetrahydrofuran (THF)/n-hexane and THF/ethyl ether respectively. Finally, the macroinitiator was dried under vacuum overnight.Preparation of P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA block copolymers
Similarily, P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA block copolymers were synthesized by ATRP of NIPAAm or DEA monomers using P(MMA-co-MPMA)-Br as the macroinitiator and CuBr/Me6TREN as the catalyst. Briefly, NIPAAm (2.26 g, 20 mmol) or DEA (2 mL, 10 mmol), macroinitiator (0.2 g, 0.04 mmol), CuBr (14.3 mg, 0.1 mmol), Me6TREN (46 mg, 0.2 mmol), DMF (2 mL) were introduced into a glass flask charged with a magnetic stirrer and degassed with N2 purge. Both polymerizations were carried out at 70 °C for 16 h. The products were purified by passing through Al2O3 column followed by repeated precipitation in ethyl ether (for P(MMA-co-MPMA)-b-PNIPAAm) or n-hexane (for P(MMA-co-MPMA)-b-PDEA) for two times. The obtained products were dried in a vacuum oven overnight.1H NMR characterization
1H NMR spectra were recorded on a Varian Unity 300 MHz spectrometer using CDCl3 and D2O as the solvents. Solid NaOH and DCl were employed as the slight additives to adjust the pH value of D2O.SEC-MALLS measurements
The size-exclusion chromatography and multi-angle laser light scattering (SEC-MALLS) analysis were used to determine the molecular weights of polymers. A dual-detector system, consisting of a MALLS device (DAWN EOS, Wyatt Technology) and an interferometric refractometer (Optilab DSP, Wyatt Technology) were used. The columns used were styragel HR1 and HR4. The injected mass for P(MMA-co-MPMA)-Br, P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA copolymers was 0.56, 0.50, and 0.58 mg, respectively. THF (chromatographic grade) was used as the eluent at a flow rate of 0.3 mL/min. The MALLS detector was operated at a laser wavelength of 690 nm. The dn/dc value of the three copolymers mentioned above was 0.103, 0.080, and 0.081 respectively.Micelle formation
P(MMA-co-MPMA)-b-PNIPAAm (3 mg) and P(MMA-co-MPMA)-b-PDEA (3 mg) were dissolved in 0.4 mL of THF, which was then added into 18 mL of aqueous solution (pH = 2, T = 20 °C) under vigorous stirring to form CCL mixed micelle. Note that the ionic strength of the aqueous solution was adjusted to 0.1 by addition of NaCl to eliminate the possible discrepancy resulting from variation of ion strength.30 After the sol–gel process (15 h), THF and byproduct methanol from the hydrolysis of the silyl ethers were removed by dialysis against distilled water for 2 days (molecular weight cut-off: 8000–12,000 g mol−1). The final product was harvested by freeze-drying before further examination.Transmission electron microscopy (TEM) observation
For aqueous phase, a drop of mixed micelle solution was placed on a copper grid with carbon film and further stained with a 0.2% (w/v) solution of phosphotungstic acid before visualization by JEM-100CX II instrument at an acceleration voltage of 100 keV. For DMF suspension, the sample was prepared in the same way, however, without staining. The specimens at high temperature were prepared according to our previous study.25 For each sample, the size of a minimum of 200 particles was measured to obtain the average size.Size distribution measurements
Nano ZS ZEN3600 (Malvern Instruments) was used to determine the average size distribution of CCL mixed micelle in aqueous solution at different temperatures and pHs. The micelle solution (polymer concentration = 0.5 g L−1, I = 0.1 M) was passed through a 0.45 μm pore-sized syringe filter (purchased from Shanghai Chemical Reagent Company) prior to measurements. Note that there are three types of intensities, namely, light, number, and volume for DLS calculation. In this study, light intensity was used.Static and dynamic laser light scattering (LLS)
A modified commercial light scattering spectrometer (ALV/SP-125, ALV, Germany) equipped with an ALV-5000/E multi-τ digital time correlator, and a He–Ne laser (at λ = 632.8 nm) was used. The details of the LLS instrumentation and theory can be found in the literature.24,31,32 Briefly, in static LLS, the angular dependence of scattered light intensity, termed the Rayleigh ratio (Rθ), of dilute polymer solution leads to the root-mean square z-average radius of gyration 〈s2〉1/2(or written as 〈Rg〉) via a Zimm plot. In dynamic LLS, the precisely measured intensity-intensity time correlation function G(2)(q,τ) in the self-beating mode can be related to the linewidth distribution G(Γ) by using the CONTIN Laplace inversion algorithm in the correlator. For a pure diffusive relaxation, G(Γ) can be converted to a translational diffusion coefficient distribution G(D) by Γ = Dq2 or a hydrodynamic radius distribution f(Rh) using the Stokes-Einstein equation 〈Rh〉 = kBT/6πηD, where kB is the Boltzmann constant, T is the absolute temperature, η is the solvent viscosity and D is the translational diffusion coefficient.LCST behaviors
The optical absorbances of P(MMA-co-MPMA)-b-PNIPAAm micelle, P(MMA-co-MPMA)-b-PDEA micelle and CCL mixed micelle in weak acid aqueous solutions at various temperatures were measured at 500 nm using a Lambda Bio40 UV-Vis spectrometer (Perkin-Elmer), respectively. The concentration for the three samples was kept the same as 0.35 g L−1. Sample cells were thermostated in a circulator bath at different temperatures from 20 to 48 °C prior to the measurements, and the heating rate was set at 0.1 °C min−1. The LCST was defined as the temperature producing a half increase of the total increase in optical absorbance.Results and discussion
Synthesis of P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA copolymers
Herein, a two-step approach was used to prepare P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA diblock copolymers, the P(MMA-co-MPMA) random copolymer was synthesized in DMF at 70 °C using CuBr/bpy as the catalyst and EBB as the ATRP initiator, and the obtained P(MMA-co-MPMA)-Br was then employed as a macroinitiator for the ATRP of NIPAAm or DEA in DMF at 70 °C using more active CuBr/Me6TREN as the catalyst (Scheme 1).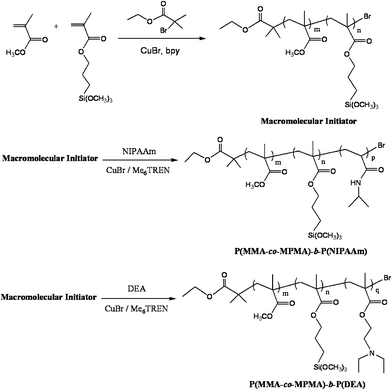 | ||
| Scheme 1 Synthesis of P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA diblock copolymers. | ||
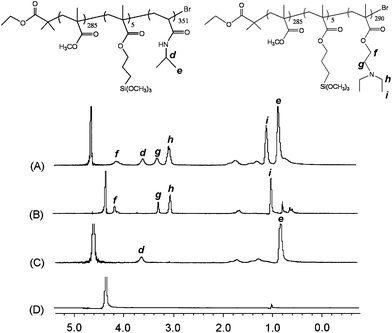
1H NMR was employed to characterize the structure of the prepared block copolymers. Fig. 2(A) showed the 1H NMR spectrum of P(MMA-co-MPMA) in CDCl3. The molar ratio of MMA and MPMA units in the P(MMA-co-MPMA) block was determined to be 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 based on the integral ratio of resonance signals at 3.6 ppm (a) and 3.5 ppm (b), characteristic peaks of MMA (-OCH3) and MPMA (-(OCH3)3) units, respectively. In comparison to Fig. 2(A), we found that the characteristic peaks of PNIPAAm (5.9–7.0 ppm (c, -NH-), 4.0 ppm (d, -CH(CH3)2), 1.0 ppm (e, -CH(CH3)2)) and PDEA (4.0 ppm (f, -OCH2-), 2.7 ppm (g, -OCH2CH2-), 2.5 ppm (h, -N(CH2CH3)2), 1.1 ppm (i, -N(CH2CH3)2)) blocks also appeared in the 1H NMR spectra of the purified P(MMA-co-MPMA)-b-PNIPAAm (Fig. 2(B)) and P(MMA-co-MPMA)-b-PDEA (Fig. 2(C)), confirming the successful synthesis of the block copolymers. However, the invisible resonance signals of MPMA unit were probably due to its rather low content in the block copolymer.
:1 based on the integral ratio of resonance signals at 3.6 ppm (a) and 3.5 ppm (b), characteristic peaks of MMA (-OCH3) and MPMA (-(OCH3)3) units, respectively. In comparison to Fig. 2(A), we found that the characteristic peaks of PNIPAAm (5.9–7.0 ppm (c, -NH-), 4.0 ppm (d, -CH(CH3)2), 1.0 ppm (e, -CH(CH3)2)) and PDEA (4.0 ppm (f, -OCH2-), 2.7 ppm (g, -OCH2CH2-), 2.5 ppm (h, -N(CH2CH3)2), 1.1 ppm (i, -N(CH2CH3)2)) blocks also appeared in the 1H NMR spectra of the purified P(MMA-co-MPMA)-b-PNIPAAm (Fig. 2(B)) and P(MMA-co-MPMA)-b-PDEA (Fig. 2(C)), confirming the successful synthesis of the block copolymers. However, the invisible resonance signals of MPMA unit were probably due to its rather low content in the block copolymer.
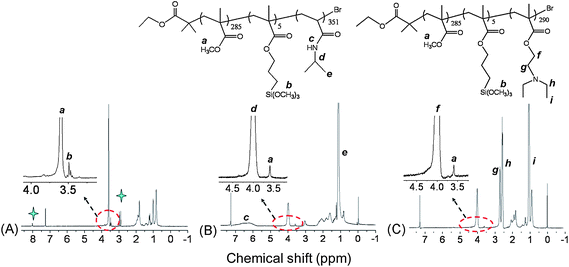 | ||
| Fig. 2 1H NMR spectra of (A) P(MMA285-co-MPMA5), (B) P(MMA285-co-MPMA5)-b-PNIPAAm351 and (C) P(MMA285-co- MPMA5)-b-PDEA290 in CDCl3. (The blue star in (A) is ascribed to the residual DMF.) | ||
SEC-MALLS were further utilized to determine the molecular weights of the as-prepared copolymers. It can be found from Table 1 that Mn of P(MMA-co-MPMA)-b-PNIPAAm (67,100 Da) and P(MMA-co-MPMA)-b-PDEA (80,700 Da) were both larger than that of P(MMA-co-MPMA) (27,090 Da). MMA, MPMA and DEA exhibited similar activity in the polymerization because they were all methacrylate monomers, as a result, the molecular weight distribution (Mw/Mn) of P(MMA-co-MPMA)-b-PDEA block copolymer was bigger than its precursor of P(MMA-co-MPMA) as expected. However, NIPAAm, as an acrylamide, possessed different polymerizable property, which possibly accounted for the decreased Mw/Mn value after its polymerization. In addition, the variation of Mw/Mn value after the second-step ATRP was also confirmed by the SEC traces presented in Fig. S1.† In comparison to the SEC trace obtained for the P(MMA-co-MPMA) random copolymer precursor, there were clear shifts to the higher molecular weights for the P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA diblock copolymers (Fig. S1†), indicating a high initiating efficiency of the P(MMA-co-MPMA)-Br macroinitiator for the polymerization of NIPAM or DEA, and a successful preparation of the target diblock copolymers by consecutive ATRP. The appearance of a small peak at elution time 24 min in the SEC trace of P(MMA-co-MPMA) was feasibly attributed to the existence of oligo(methyl methacrylate) (OMMA). Although the product P(MMA-co-MPMA) has been purified by repeated precipitation, it remained difficult to get rid of OMMA completely possibly due to their similar properties. However, this small peak was also observed in the SEC traces of both block copolymers, indicating that OMMA exhibited no initiating ability for the further polymerization, therefore the effect of such impurity on the properties of the final diblock copolymers was negligible. The degrees of polymerization (DP) of the MMA and MPMA units were calculated to be 285 and 5 according to the molar ratio of these two units (57:1) and the molecular weight of P(MMA-co-MPMA) block (27,090 Da). Then the DP of PNIPAAm and PDEA blocks were estimated to be 351 and 290 from the corresponding molecular weight (67,100 Da and 80,700 Da) of the two diblock copolymers, respectively. The diblock copolymers were thus denoted P(MMA285-co-MPMA5)-b-PNIPAAm351 and P(MMA285-co-MPMA5)-b-PDEA290. It should be pointed out that the actual composition of the copolymer can be calculated based on either the integrity ratio in the NMR spectra or the absolute molecular weight determined by SEC-MALLS. Both methods were used in our experiment, however, the results obtained from molecular weight were closer to the molar feed ratio of the copolymer, therefore we, herein, only report the calculation of the real composition using the data from molecular weight.
| Copolymer | M n (Da) | M w/Mn |
|---|---|---|
| P(MMA-co-MPMA) | 27,090 | 1.61 |
| P(MMA-co-MPMA)-b-PNIPAAm | 67,100 | 1.37 |
| P(MMA-co-MPMA)-b-PDEA | 80,700 | 1.85 |
CCL mixed micelle formation
The morphology of the resulting CCL mixed micelles was investigated by TEM and DLS. A typical photograph of the CCL mixed micelles at pH 5, 20 °C was shown in Fig. 3(A1), in which the CCL mixed micelles clearly displayed a well-defined spherical shape with size around 70–140 nm in diameter. As shown in Fig. 3(B1), the CCL mixed micelles exhibited narrow size distribution with an average diameter of around 210 nm. Compared with the diameter obtained by TEM, the typically enlarged size determined by dynamic light scattering (DLS) was attributed to the fact that what was measured by DLS was the hydrodynamic diameter in an aqueous medium and reflected the dimensions of both compacted core and stretched shell, whereas the TEM observation revealed the conformation in the dry state.33,34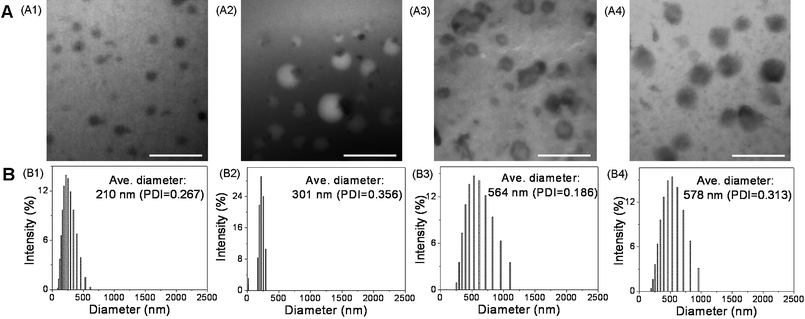 | ||
| Fig. 3 (A) TEM images and (B) size distributions of CCL mixed micelles: (A1, B1) at pH 5, 20 °C; (A2, B2) in DMF; (A3, B3) at pH 5, 45 °C; (A4, B4) at pH 9, 20 °C. Scale bars indicate 700 nm. | ||
After cross-linking, the lyophilized CCL mixed micelles were dissolved in an organic solvent to prove sufficient cross-linking and further explore their shape persistence against alteration of solvents. Fig. 3(A2) displayed the TEM image obtained for the CCL mixed micelles in DMF, which convincingly showed that the cross-linked structure of the mixed micelle was still after transfer into the organic medium, in which the assembly would be destroyed. After switching the solvent from water to DMF, the CCL mixed micelles maintained their integrity and still exhibited well-defined spherical shapes with diameters ranging from 130 to 390 nm. The structure coincided with the morphology observed in the aqueous phase (Fig. 3(A1)). It turned out that cross-linking resulting from the hydrolysis and condensation of trimethoxysilyl functions was effective and the mixed micellar structure was locked by cross-linking of the hydrophobic core. Similar results were reported in our previous studies23,24 as well.
As shown in Fig. 3(B2), the average size of the CCL mixed micelles was around 301 nm, which agreed roughly with the value observed by TEM. In addition, upon comparison of part B2 and B1 in Fig. 3, we found that the size of the CCL mixed micelle (301 nm) recorded in DMF was larger than that measured in water (210 nm). The reason was probably that DMF was capable of dissolving P(MMA-co-MPMA), PNIPAAm and PDEA blocks; hence, both the core and shell of the CCL mixed micelles were fully solvated and swelled in DMF. However, the use of water as the solvent resulted in insufficient mobility and contraction of PMMA block, leading to a smaller size.
Thermo and pH double responsive properties of CCL mixed micelles
The resultant CCL mixed micelles were anticipated to undergo thermo- and pH- double responsive aggregation behaviors due to the combination of separate responsive properties exhibited by the two block copolymers. Absorbance measurements, TEM, LLS and 1H NMR were utilized to investigate the dual responsiveness of the CCL mixed micelle in aqueous solution.From the optical absorbance of the micelle aqueous solution as a function of temperature (Fig. 4), we found that the P(MMA-co-MPMA)-b-PNIPAAm micelle (Fig. 4(A)) underwent a change in its structure at the temperature corresponding to the LCST of PNIPAAm homopolymer, ca. 33 °C. The results confirmed that the completely phase-separated core-shell micellar structure led to the unaltered LCST behavior of the shell-forming block. However, the absorbance of P(MMA-co-MPMA)-b-PDEA micelle kept constant with increasing temperature (Fig. 4(C)), indicating that the P(MMA-co-MPMA)-b-PDEA micelle was stable against temperature and exhibited no thermo-sensitivity. Most importantly, the CCL mixed micelle (Fig. 4(B)) constructed from the coaggregation of the two block copolymers exhibited almost the same LCST behaviors (LCST value and response rate) as the P(MMA-co-MPMA)-b-PNIPAAm micelle, suggesting that the PDEA chains had little effect on the thermo-responsiveness of the PNIPAAm chains in the dual responsive shells of the mixed micelle, therefore we concluded that the desired CCL mixed micelle with unaltered thermo-sensitivity was developed.
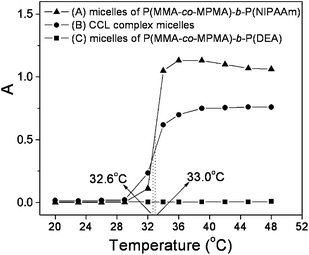 | ||
| Fig. 4 Thermo-sensitive behaviors of micelle solution at pH 5: (A) P(MMA285-co-MPMA5)-b-PNIPAAm351 micelles, (B) CCL mixed micelles, and (C) P(MMA285-co-MPMA5)-b-PDEA290 micelles. | ||
The lyophilized CCL mixed micelles were re-dissolved in different aqueous media for morphology observation to further confirm their dual sensitivity. TEM images of the CCL mixed micelles revealed the presence of well-dispersed spherical micelles of around 100–240 and 110–310 nm in diameter at pH 5, 45 °C (Fig. 3(A3)) and pH 9, 20 °C (Fig. 3(A4)), respectively. It was worth pointing out that for the individual P(MMA-co-MPMA)-b-PNIPAAm or P(MMA-co-MPMA)-b-PDEA micelles, large aggregates were believed to precipitate from the micelle solution when the temperature increased above the LCST of PNIPAAm or pH rose above the pKa of PDEA due to the insolubility of PMMA, PNIPAAm (at temperature higher than its LCST) and PDEA (at alkaline pH) chains. However, stable mixed micelles with regularly shape were observed irrespective of increased temperature (Fig. 3(A3)) or pH value (Fig. 3(A4)) under TEM visualization, which was attributed to the presence of PNIPAAm/PDEA dual shells in the structure of the mixed micelles, that is, the hydrophilic PDEA or PNIPAAm chains can still stabilize the micelles and prevent the formation of aggregates at pH 5 or 20 °C respectively. The results further confirmed the formation of mixed micelles at the molecular level from the mixture of P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA. The particle sizes estimated from TEM were systematically smaller than those measured by DLS, which were ∼564 and 578 nm in diameter for the CCL mixed micelles at pH 5, 45 °C (Fig. 3(B3)) and pH 9, 20 °C (Fig. 3(B4)), respectively. In comparison to the data at pH 5, 20 °C (Fig. 3(A1), we found that the mixed micelles became larger when subjected to high temperature (45 °C, Fig. 3(A3)) or alkaline pH (pH 9, Fig. 3(A4)). The reason was probably that the structural transformation of PNIPAAm or PDEA chains from the stretching state as the hydrophilic shells to the contracting status as the hydrophobic inner shells increased the hydrophobic domain in the micellar structure,35,36 since the hydrophobic region was emphasized after negative staining in TEM specimen,37 the mixed micelles with enlarged size were visualized consequently.
〈Rg〉 and 〈Rh〉 of the CCL mixed micelle under different conditions were further determined by LLS analyses. It can be seen from Table 2 that the 〈Rg〉 and 〈Rh〉 recorded at high temperature (45 °C, pH 5) or alkaline pH (25 °C, pH 9) were both larger as compared with the data measured at room temperature and acidic pH (25 °C, pH 5). The increase of micelle dimension observed in LLS study agreed well with the results of TEM and size distribution measurements, also confirming the occurrence of structural transformation in the mixed shells of micelle at elevated temperature or pH value. In addition, the 〈Rg〉/〈Rh〉 value of the mixed micelles was determined to be around 0.7 under different conditions, close to the theoretical calculated value for a uniform sphere (∼0.8),24,38 indicating a spherical shape as well as ill-uniform core-shell structure for the CCL mixed micelle.
Fig. 5 shows the temperature- and pH-dependent 1H NMR spectra for the CCL mixed micelles recorded in D2O. Note that the proton signals from PMMA chains were invisible because they formed the immobile and non-solvated micellar core in D2O. At pH 5 and 20 °C (Fig. 5(A)), all the characteristic signals of PNIPAAm (d and e) and PDEA (f, g, h and i) blocks were readily visible, indicating that both blocks were hydrophilic and molecularly solvated. Increasing the temperature to 45 °C at pH 5 (Fig. 5(B)) led to the hydrophobic collapse of the stretched PNIPAAm chain from an extended-coil conformation to a shrunken-globule conformation, as evidenced by the significant attenuation of peaks d and e associated with PNIPAAm block, however, the characteristic resonances of the PDEA block were clearly evident, which was reasonable as this block was soluble at pH 5 due to the protonation of tertiary amine residues. Based on our assumption, CSC mixed micelles consisting of a cross-linked PMMA core, PNIPAAm inner shell, and protonated PDEA outer corona formed at pH 5 and 45 °C (Fig. 6).
 | ||
| Fig. 5 1H NMR spectra of CCL mixed micelles in D2O: (A) pH 5, 20 °C; (B) pH 5, 45 °C; (C) pH 9, 20 °C; (D) pH 9, 45 °C. | ||
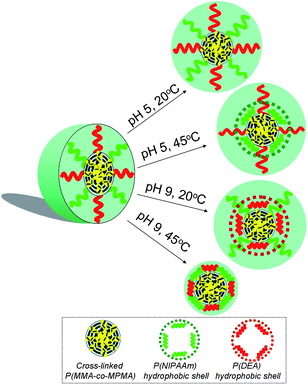 | ||
| Fig. 6 Schematic illustration of temperature and pH double responsive structural changes for CCL mixed micelle. | ||
Conversely, upon adjusting to pH 9 and 20 °C (Fig. 5(C)), the stretched PDEA chains collapsed due to their deprotonation, leading to the complete disappearance of characteristic peaks of PDEA chains, while the signals of PNIPAAm chains were still clearly visible because of their hydrophilicity at temperatures below the LCST. The results indicated the formation of three-layer micelle structure with cross-linked PMMA core, hydrophobic PDEA inner shell, and hydrophilic PNIPAAm outer corona at pH 9 and 20 °C (Fig. 6).
In addition, starting from the state at pH 5 and 20 °C, all the characteristic peaks of PNIPAAm and PDEA blocks were hardly detected at pH 9 and 45 °C, further confirming the occurrence of transformation from the hydrophilic to hydrophobic state for both blocks at high temperature and pH value (Fig. 6).
From the above discussion, we concluded that the CCL core-shell mixed micelles self-assembled from the mixtures of P(MMA285-co-MPMA5)-b-PNIPAAm351 and P(MMA285-co-MPMA5)-b-PDEA290 converted into CSC mixed micelles at elevated temperature or pH value. The resultant CSC mixed micelle possessed a three-layered structure with the hydrophobic PMMA block as the compacted core surrounded by the collapsed PNIPAAm blocks as the inner shell and soluble PDEA chains as the outer corona at high temperature, or shrunk PDEA blocks as the inner shell and stretched PNIPAAm chains as the outer corona at alkaline pH. In addition, it is worth pointing out that the transformation of both shell-forming blocks was reversible as demonstrated by the transition from a turbid solution to a transparent solution when the aqueous temperature was decreased from above (45 °C) to below (20 °C) the LCST or the aqueous pH shifted from alkaline (pH 9) to acidic (pH 5) value.
Conclusion
Two well-defined stimulus-responsive P(MMA-co-MPMA)-b-PNIPAAm and P(MMA-co-MPMA)-b-PDEA block copolymers with the same hydrophobic block were synthesized via successive ATRP. CCL mixed micelle was prepared by co-self-assembly of the two block copolymers in acid aqueous solution at room temperature. A combination of analytical techniques (absorbance, TEM, LLS and 1H NMR) revealed the conversion of core-shell mixed micelles into CSC mixed micelles at elevated temperature or pH value. Most importantly, the size of the CCL mixed micelles could be readily regulated by adjusting the environmental conditions (temperature and/or pH) due to the existence of thermo- and pH- dually responsive shells composed of PNIPAAm and PDEA. The CCL mixed micelles developed herein would have great potential to be novel carriers for both temperature and pH controlled drug delivery.Acknowledgements
We acknowledge the financial support from the Ministry of Science and Technology of China (2009CB930300), Trans(New)-Century Training Programme Foundation for the Talents from the Ministry of Education of China and Natural Science Foundation of Hubei Province, China (2009CDA024).References
- M. Oishi, T. Hayama, Y. Akiyama, S. Takae, A. Harada, Y. Yamasaki, F. Nagatsugi, S. Sasaki, Y. Nagasaki and K. Kataoka, Biomacromolecules, 2005, 6, 2449–2454 CrossRef CAS.
- S. Chosh, K. Irvin and S. Thayumanavan, Langmuir, 2007, 23, 7916–7919 CrossRef CAS.
- S. Fujii, Y. Cai, J. V. M. Weaver and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 7304–7305 CrossRef CAS.
- J. Huang, L. Ren and Y. Chen, Polym. Int., 2008, 57, 714–721 CrossRef CAS.
- M. G. J. T. Cate and H. G. Börner, Macromol. Chem. Phys., 2007, 23, 11866–11874.
- H. Wei, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- X. Xu, A. E. Smith, S. E. Kirkland and C. L. McCormick, Macromolecules, 2008, 41, 8249–8435.
- Y. H. Hsu, W. H. Chiang, M. C. Chen, C. S. Chern and H. C. Chiu, Langmuir, 2006, 22, 6764–6770 CrossRef CAS.
- J. C. Leroux, E. Roux, D. L. Garrec, K. Hong and D. C. Drummond, J. Controlled Release, 2001, 72, 71–84 CrossRef CAS.
- S. A. Angelopoulos and C. Tsitsilianis, Macromol. Chem. Phys., 2006, 207, 2188–2194 CrossRef CAS.
- X. Jiang, Z. Ge, J. Xu, H. Liu and S. Y. Liu, Biomacromolecules, 2007, 8, 3184–3192 CrossRef CAS.
- H. Wei, X. Z. Zhang, H. Cheng, W. Q. Chen, S. X. Cheng and R. X. Zhuo, J. Controlled Release, 2006, 116, 266–274 CrossRef CAS.
- T. Qu, A. Wang, J. Yuan and Q. Gao, J. Colloid Interface Sci., 2009, 336, 865–871 CrossRef CAS.
- C. L. Lo, C. K. Huang, K. M. Lin and G. H. Hsiue, Biomaterials, 2007, 28, 1225–1235 CrossRef CAS.
- D. Xiong, Y. An, Z. Li, R. Ma, Y. Liu, C. Wu, L. Zou, L. Shi and J. Zhang, Macromol. Rapid Commun., 2008, 29, 1895–1901 CrossRef CAS.
- D. Xiong, Z. Li, L. Zou, Z. He, Y. Liu, Y. An, R. Ma and L. Shi, J. Colloid Interface Sci., 2010, 341, 273–279 CrossRef CAS.
- Y. Zhuang, J. P. Lin, L. Q. Wang and L. S. Zhang, J. Phys. Chem. B, 2009, 113, 1906–1913 CrossRef CAS.
- S. H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2009, 42, 25–29 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- Y. Shen, S. Zhu and R. Pelton, Macromolecules, 2001, 34, 3182–3185 CrossRef CAS.
- L. Ren, F. Ke, Y. Chen, D. Liang and J. Huang, Macromolecules, 2008, 41, 5295–5300 CrossRef CAS.
- G. Zheng and H. D. H. Stover, Macromolecules, 2002, 35, 7612–7619 CrossRef CAS.
- H. Wei, C. Cheng, C. Chang, W. Q. Chen, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Langmuir, 2008, 24, 4564–4570 CrossRef CAS.
- H. Wei, D. Q. Wu, Q. Li, C. Chang, J. P. Zhou, X. Z. Zhang and R. X. Zhuo, J. Phys. Chem. C, 2008, 112, 15329–15334 CrossRef CAS.
- C. Chang, H. Wei, J. Feng, Z. C. Wang, X. J. Wu, D. Q. Wu, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Macromolecules, 2009, 42, 4838–4844 CrossRef CAS.
- G. Li, L. Guo, S. Ma and J. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1804–1810 CrossRef CAS.
- K. Zhang, L. Gao and Y. Chen, Macromolecules, 2007, 40, 5916–5922 CrossRef CAS.
- Y. Zhang, W. Gu, H. Xu and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2379–2389 CrossRef CAS.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1996, 5, 41–44.
- X. Wang, H. Tu, P. V. Braun and P. W. Bohn, Langmuir, 2006, 22, 817–823 CrossRef CAS.
- Y. Jin, L. N. Zhang, M. Zhang, L. Chen, P. C. K. Cheung, V. E. C. Oi and Y. Lin, Carbohydr. Res., 2003, 338, 1517–1521 CrossRef CAS.
- Z. C. Ma, J. G. Wang and L. N. Zhang, Biopolymers, 2008, 89, 614–622 CrossRef CAS.
- W. J. Liang, C. H. Lien and P. L. Kuo, J. Colloid Interface Sci., 2006, 294, 371–375 CrossRef CAS.
- C. Chang, H. Wei, C. Y. Quan, Y. Y. Li, J. Liu, Z. C. Wang, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3048–3057 CrossRef CAS.
- S. D. Santis, R. D. Ladogana, M. Diociaiuti and G. Masci, Macromolecules, 2010, 43, 1992–2001 CrossRef CAS.
- D. Xiong, Z. He, L. An, Z. Li, H. Wang, X. Chen and L. Shi, Polymer, 2008, 49, 2548–2552 CrossRef CAS.
- J. Madsen, S. P. Armes, K. Bertal, H. Lomas, S. MacNeil and A. L. Lewis, Biomacromolecules, 2008, 9, 2265–2275 CrossRef CAS.
- A. Z. Niu, D. J. Liaw, H. C. Sang and C. Wu, Macromolecules, 2000, 33, 3492–3494 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEC traces of P(MMA285-co-MPMA5), P(MMA285-co-MPMA5)-b-PNIPAAm351, and P(MMA285-co- MPMA5)-b-PDEA290. See DOI: 10.1039/c0py00373e |
| ‡ Both authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2011 |