DOI:
10.1039/C0PY00372G
(Paper)
Polym. Chem., 2011,
2, 815-822
Received
16th November 2010
, Accepted 15th December 2010
First published on 24th January 2011
Introduction
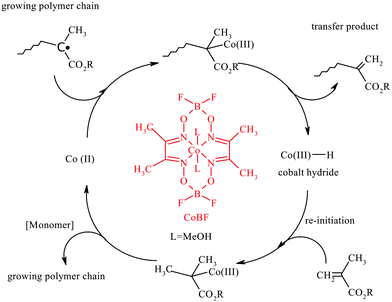
Catalytic chain transfer polymerization is a very efficient and versatile free-radical polymerization technique for the synthesis of macromonomers with ω-unsaturated vinyl terminal groups.1,2 It is used commercially by DSM and DuPont for the production of viscosity modifiers and engine additives for the automotive industry. The reaction is mediated by the ability of certain low-spin Co(II) complexes, such as cobaloximes, to efficiently catalyze the chain transfer to monomer reaction.3 A widely investigated Co(II) transfer agent is bis(boron difluorodimethylglyoximate) cobalt(II) (CoBF).1,2,4–6 The chain transfer constant of CoBF is typically 102 to 104 for monomers containing an α-methyl group, such as methacrylates. CCT occurs via a mechanism characterized by two consecutive steps:3,7 firstly, the Co(II) complex abstracts a hydrogen from the propagating radical to form a dead polymer chain and a Co(III)–H complex; then this complex reacts with another monomer molecule to produce the original Co(II) complex and another propagating radical (Scheme 1). The Co(II) complex acts in a catalytic fashion and therefore is not consumed in the process. CCT is a very powerful technique used to synthesize low molecular weight polymers/oligomers terminated by a vinyl end-group (macromonomers) with a large range of functional or non-functional monomers. CCT polymerization can be performed in a large range of solvents, ranging from aqueous to nonpolar organic media.8,9
Inspired by the work reported by Ishizone et al.10 and later by Lutz and Hoth11 and others, the preparation of thermoresponsive, antifouling PEG based polymers is of huge interest for the preparation of thermoresponsive micelles, hybrid organic/inorganic nanoparticles,12–14 surface modification,15–18 hyperbranched/nanogels and protein–polymer conjugates.19–25 As illustrated by the recent publication of Dong and Matyjaszewski who describe the first preparation of thermally responsive poly(oligo(ethylene glycol) methyl ether methacrylate) via AGET ATRP in miniemulsion.26 In this current study, the synthesis of thermoresponsive PEG based macromonomers by the copolymerization of OEGMEMA and DEGMEMA using CCT is described. The lower critical solution temperature (LCST) was tuned by the composition of these two monomers to yield thermoresponsive polymers with LCSTs ranging from 30 to 90 °C. To the best of our knowledge, this is the first preparation of thermally responsive poly(oligo(ethylene glycol) methyl ether methacrylate) via Catalytic Chain Transfer Polymerization. The advantage of CCT is that the resulting polymer chains have unsaturated vinylic end-groups (with very high chain-end fidelity; close to 100%) which create a large range of synthetic options for end-group modification with simple organic compounds. Over the last decade, “click” chemistry has opened new avenues for the design and creation of tailor-made macromolecules.27–29 Thiol–ene “click” chemistry, in which thiols react quantitatively with double bonds, has been shown to be highly efficient in a large range of solvents, without the addition of metals, under mild conditions.30–38 There are two types of thiol–ene reaction: (i) a base catalyzed Michael addition and (ii) the anti-Markovnikov radical UV initiated click. Both of these approaches have been used for the functionalization of polymers31,39 or for the design of complex macromolecules.40–45
In this study, thermoresponsive PEG-based copolymers and homopolymers of OEGMEMA obtained by CCT were modified using thiol–ene click chemistry with a variety of different functional thiol compounds to yield functional thermoresponsive polymers in high yield. The optimal conditions for thiol–ene click chemistry to obtain a very high degree of functionality (solvent and nature of the catalyst) are reported. The polymers were rigorously characterized via electrospray mass spectrometry (ESI-MS), 1H NMR spectroscopy and MALDI-ToF MS analysis. In addition, the thermoresponsive properties (LCST) and thermal properties (glass transition and decomposition temperatures) of the copolymers were determined before and after thiol–ene modification.
Experimental
Materials
The CCT agent bis(methanol) complex, (CH3OH)2Co–(dmgBF2)2 (CoBF), was synthesized according to the method of Bakac et al.46,47 The CTA transfer efficiency was checked by CCT polymerization of methyl methacrylate (MMA) which yielded a CS value = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The monomers, OEGMEMA475 (Aldrich, 95%), DEGMEMA (Aldrich, 95%), and OEGMEMA1100 (Aldrich, 95%), were used as received. The initiator 4,4′-azobis(4-cyanovaleric acid) (ACVA) was used as received and 2,2′-azobisisobutyronitrile (AIBN) was purified by recrystallisation twice from methanol. The thiols, 2-mercaptoethanol (2-ME, 99%, Sigma-Aldrich), 1-dodecanethiol (DT, 98+%, Sigma-Aldrich), benzyl mercaptan (BM, 99%, Sigma-Aldrich), 3-mercaptopropionic acid (3-MPA, 99%, Sigma-Aldrich), triethylamine (TEA, 99%, Fisher Scientific UK Ltd.), hexylamine (HA, 99%, Sigma-Aldrich), and dimethylphenylphosphine (DMPP, 99%, Aldrich) were used as received.
General catalytic chain transfer polymerizations (CCTPs) were performed as follows: monomers (DEGMEMA, OEGMEMA475, and OEGMEMA1100) and solvents (acetonitrile or water/methanol mixture) were purged with N2 gas for at least an hour prior to use. Stock solutions of CoBF were also weighed and purged with N2 prior to use. AIBN or CVA was weighed into Schlenk flasks. All reaction mixtures were then mixed under N2 gas. To ensure the absence of O2, the reaction mixtures were subjected to at least three freeze–pump–thaw cycles. Polymerizations were performed at a constant temperature of 70 °C. Polymerizations were stopped by cooling and subsequent exposure to air. Polymers were then purified by precipitation and dialysis. Poly(OEGMEMA) was precipitated in diethyl ether and poly(DEGMEMA) was precipitated in water or cold petroleum ether (40–60 °C).
000. The monomers, OEGMEMA475 (Aldrich, 95%), DEGMEMA (Aldrich, 95%), and OEGMEMA1100 (Aldrich, 95%), were used as received. The initiator 4,4′-azobis(4-cyanovaleric acid) (ACVA) was used as received and 2,2′-azobisisobutyronitrile (AIBN) was purified by recrystallisation twice from methanol. The thiols, 2-mercaptoethanol (2-ME, 99%, Sigma-Aldrich), 1-dodecanethiol (DT, 98+%, Sigma-Aldrich), benzyl mercaptan (BM, 99%, Sigma-Aldrich), 3-mercaptopropionic acid (3-MPA, 99%, Sigma-Aldrich), triethylamine (TEA, 99%, Fisher Scientific UK Ltd.), hexylamine (HA, 99%, Sigma-Aldrich), and dimethylphenylphosphine (DMPP, 99%, Aldrich) were used as received.
General catalytic chain transfer polymerizations (CCTPs) were performed as follows: monomers (DEGMEMA, OEGMEMA475, and OEGMEMA1100) and solvents (acetonitrile or water/methanol mixture) were purged with N2 gas for at least an hour prior to use. Stock solutions of CoBF were also weighed and purged with N2 prior to use. AIBN or CVA was weighed into Schlenk flasks. All reaction mixtures were then mixed under N2 gas. To ensure the absence of O2, the reaction mixtures were subjected to at least three freeze–pump–thaw cycles. Polymerizations were performed at a constant temperature of 70 °C. Polymerizations were stopped by cooling and subsequent exposure to air. Polymers were then purified by precipitation and dialysis. Poly(OEGMEMA) was precipitated in diethyl ether and poly(DEGMEMA) was precipitated in water or cold petroleum ether (40–60 °C).
For semi-batch polymerizations, the procedure was the same, however, the monomer and CoBF solutions were added at a rate of 1 mL min−1 throughout the reactions.
General CCT copolymerization procedure was as follows: monomers OEGMEMA475 and DEGMEMA with different compositions and solvents (acetonitrile or water/methanol mixture) were mixed with AIBN and purged with N2 gas at least one hour before use. Stock solutions of CoBF were also prepared and purged before use. All reaction mixtures were then mixed under N2 gas. To ensure the absence of O2, the reaction mixtures were then subjected to at least three freeze–pump–thaw cycles. Polymerizations were performed at a constant temperature of 70 °C. Polymerizations were stopped by cooling and subsequent exposure to air. Copolymers were then purified by dialysis in water/acetone.
Poly(OEGMEMA) was mixed with hexylamine or triethylamine, water, acetone, acetonitrile or DMSO used as solvents, and thiol reagent. In a typical reaction, 1 eq. of poly(OEGMEMA) (100 mg), 2 eq. of thiol, and 3 eq. of hexylamine were added into a flask and then purged for 15 minutes with N2. The vials were then placed at ambient temperature or in a 40 °C oil bath for 14 hours before the reaction was stopped. The reaction mixtures were purified via precipitation in cold petroleum ether or dialysis.
DMPP catalyzed thio-click reactions
Polymers, thiols and DMPP were mixed in a NMR tube containing acetone-d6, acetonitrile-d3 or DMSO-d6. The reactions were performed at room temperature and monitored by 1H NMR spectroscopy. In a typical reaction, 1 eq. of polymer (0.05 g), 1.5 eq. of thiol and 0.05 eq. (1.7 mg) of DMPP were added to a NMR tube containing 0.5 mL of deuterated solvent. The reaction was monitored by the disappearance of the vinyl bond seen at 5.5–6.0 ppm.
Characterization
Nuclear magnetic resonance (NMR).
1H NMR spectroscopy was used to determine the molecular weight and structural information for confirmation of thiol–ene click conjugation. NMR spectra were recorded on a Bruker DPX-300 MHz and Bruker DPX-400 MHz.
Cloud points measurements.
The transmittance of the solution was measured at λ = 500 nm through 10 mm path length cuvettes with a micro-stir bar and total volumes of 2 mL referenced against distilled water, using a PerkinElmer Lambda 35 UV-VIS spectrometer. The water-jacketed sample and reference cuvette holders were coupled with a Peltier PTP 1 + 1 Peltier temperature programmer adjusted at a heating rate of 1 °C min−1. The cloud points (CP) were defined as the temperature corresponding to a 50% reduction in the original transmittance of the solution.
Mass spectrometry (MALDI-ToF).
MALDI-ToF analysis was performed with a Bruker Ultraflex III equipped with a neodymium-doped yttrium aluminium garnet laser (Nd:YAG). The matrix solution was prepared by dissolving trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) in a 1 mL mixture of acetone and water (equal volumes). Sodium iodide was added at 0.1% overall concentration and the polymer dissolved to a concentration of 10 mg cm−3. The matrix solution (0.5 μL) was applied to the stainless steel side and the solvent allowed to evaporate. The same volume of the polymer sample solution was applied on top of the dried matrix, and the overall mixture allowed to dry. The sample was irradiated with 300–600 pulsed laser shots at a 1–10% laser power. Calibration was performed with various linear poly(ethylene glycol) methyl ether standards.
Results and discussion
Catalytic chain transfer polymerizations of various oligo(ethylene glycol) methyl ether methacrylates were performed using 2,2′-azobisisobutyronitrile (AIBN) as the initiator, the bis(methanol) complex, (CH3OH)2Co–(dmgBF2)2 (CoBF), as a catalyst and acetonitrile as a solvent at 70 °C. The chain transfer constant of catalytic chain transfer polymerization can be easily calculated using the modified Mayo equation (eqn (1)),1,2,48 provided the monomer conversion is maintained below 10%.| | | 1/DPn = 2 + CS × ([C]/[M]) | (1) |
where DPn is the degree of polymerization, [C] is the concentration of the catalyst, and [M] is the initial monomer concentration. The monomers that were used in this study were diethylene glycol methyl ether methacrylate, oligo(ethylene glycol) methyl ether methacrylate (475), and oligo(ethylene glycol) methyl ether methacrylate (1100) as depicted in Scheme 2.
 |
| | Scheme 2 Representation of CoBF and various oligo(ethylene glycol) monomers used in this study. | |
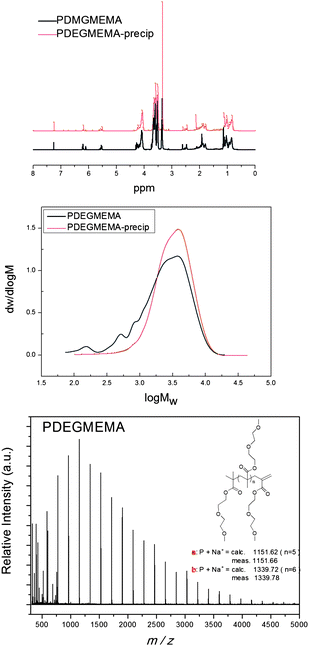
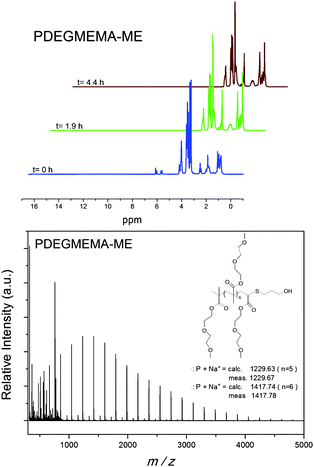
The polymer molecular weights were analyzed via GPC and 1H NMR spectroscopy. All the polymers exhibited monomodal distributions with PDI < 1.5. The polymers, after purification, were analyzed by 1H NMR and by MALDI-ToF (Fig. 1), and the molecular weights were calculated using the signal of the vinyl group at 5.5–6.0 ppm and eqn (2). It is noteworthy that the signals characteristic of the vinyl bond in the monomer and in the macromonomer are slightly different, i.e. 5.6–6.1 ppm and 5.5–6.0 ppm for monomer and macromonomer, respectively. This slight shift avoids any error in the molecular weight determination using NMR analyses (from traces of residual monomer).
| | Mn, 1H NMR = (ICH2OCO/ICH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) × MWOEGMEMA CH) × MWOEGMEMA | (2) |
where
MWOEGMEMA, I
CH2OCO and I
CHCH correspond to the molar masses of OEGMEMA, integral of the signal at 4.1 ppm (attributed to CH
2 in the adjacent position of
ester group) and of
vinyl group, respectively. The molecular weights calculated by
1H NMR were in accordance with the molecular weights determined by MALDI-ToF.
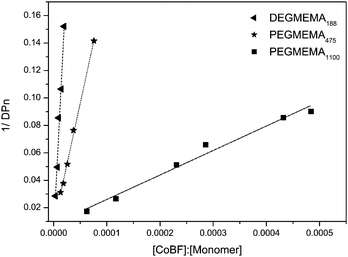
The plot of molecular weight versus the ratio of [CoBF]/[monomer] gives the chain transfer constant of CoBF for each monomer (Fig. 2).
CCTP of OEGMEMA
The chain transfer constants (CS) of CoBF to DEGMEMA, POEGMEMA475, and POEGMEMA1100 have been calculated as 7600, 1800, and 180, respectively (in acetonitrile at 70 °C). The CS values decrease proportionally with the chain length of the side chain (in this case, oligo(ethylene glycol)). A similar result was obtained by Forster and co-workers5,49 for alkyl methacrylate (methyl methacrylate, ethyl methacrylate, and butyl methacrylate). The end-group fidelity of the polymers was well maintained as confirmed by MALDI-ToF for all monomers examined. According to the concentration of CoBF, it is possible to synthesize polymer with a DPn from 7 to 50 in acetonitrile with a PDI less than 1.5 (see earlier comments on PDI veracity).
As OEGMEMA and CoBF are water soluble, it was decided to investigate the polymerization of these monomers in water at 80 °C. The addition of methanol (50 v%), as a co-solvent, was successful for the polymerization of DEGMEMA {LCST ≈ 26 °C}. Table 1 shows the evolution of the molecular weights and PDI obtained for the polymerizations of OEGMEMA and DEGMEMA in water or water/methanol. For polymerization of OEGMEMA in water, poor control of the polymerization was observed as shown by a broadening of the molecular weight distributions. In comparison when acetonitrile was used as a solvent, narrower distributions were obtained. In addition, the molecular weights obtained in acetonitrile were usually lower than the molecular weights obtained in water, showing a better efficiency of transfer. It is possible that this difference in transfer efficiency could be attributed to the slight difference of temperature. However, Davis and Kukulj50 and Heuts et al.51 show that the efficiency of CoBF (or the transfer constant) is relatively unaffected by temperature. An increase in the temperature causes an increase in propagation rate (kp) as well as an increase in transfer rate (ktr), resulting in a null effect on the CS (CS = kp/ktr). Another possible explanation is that the differences reported here are attributable to solvent effects. Haddleton and co-workers52 have previously reported that CoBF can be degraded in water at elevated temperatures (∼70 °C) and that the kinetics of the degradation process can be accelerated at low pH values. The degradation of CoBF causes a decrease in efficiency of the catalyst, inducing significant increases in the PDI during the polymerization. In the case of polymerization with CoBF in water, a semi-batch approach is necessary to keep the concentration of the CoBF constant thereby maintaining lower PDI.52 As expected, improved PDIs and control of molecular weights were observed for polymers obtained using a feed methodology compared to a batch approach (Table 1, runs P4–P7). In the batch approach, the CoBF is degraded during the polymerization, resulting in a change of the [M]/[CoBF] ratio and a subsequent drift in the polymer chain distribution.
| Run |
Monomer |
CoBF/ppm |
Technique |
M
n, GPC/g mol−1 |
PDI |
|
Polymerization was carried out in mixture solvents: methanol/water (50/50 v%).
|
| P1a |
DEGMEMA |
50 |
Feed |
11000 |
1.39 |
| P2a |
DEGMEMA |
100 |
Feed |
4600 |
1.31 |
| P3a |
DEGMEMA |
100 |
Batch |
4600 |
1.32 |
| P4 |
OEGMEMA475 |
50 |
Feed |
3200 |
1.68 |
| P5 |
OEGMEMA475 |
100 |
Feed |
2600 |
1.31 |
| P6 |
OEGMEMA475 |
50 |
Batch |
2300 |
1.85 |
| P7 |
OEGMEMA475 |
100 |
Batch |
4500 |
2.03 |
Inspired by the work of Lutz et al.11,53 and Ishizone et al.,10 the copolymerization of oligo(ethylene glycol) methyl ether methacrylate (OEGMEMA475) with diethylene glycol methyl ether methacrylate (DEGMEMA) using CCT to yield thermoresponsive macromonomers was investigated. Copolymerizations were performed in acetonitrile at 70 °C for 14 hours. The feed compositions of these monomers were varied from 50/50 to 90/10 mol% of OEGMEMA/DEGEMMA. The final composition of these copolymers was calculated from 1H NMR using the ratio between –OCH2 (ether group of OEG or DEG chains) and OCH3 (from end-group). Table 2 shows the final composition of each copolymer (F). It is interesting to note that the monomer feed ratio and the final copolymer composition do not differ significantly at low conversion indicative that the reactivity ratios of the monomers are similar, implying that compositional drift is not a major factor in the reactions. The presence of composition drift during copolymerization can result in the synthesis of gradient copolymers, when a living radical polymerization is used,54 or in the synthesis of heterogeneous copolymers, when a non-living free-radical polymerization is employed (such as CCTP).55
| Run |
f (initial feed) |
F (final composition) |
M
n, GPC/g mol−1 |
M
n, NMR/g mol−1 |
| CP1 |
50:50 |
48:52 |
2800 |
3000 |
| CP2 |
70:30 |
69:31 |
2600 |
2700 |
| CP3 |
80:20 |
76:24 |
2600 |
3200 |
| CP4 |
90:10 |
92:8 |
3000 |
3400 |
The copolymer composition of DEGMEMA and OEGMEMA allows tuning of the lower critical solution temperatures (LCSTs) from 30 to 90 °C as determined by turbidimetry measurements. Fig. 3 shows the evolution of LCST with copolymer composition. An increase of DEGMEMA in the copolymers causes a decrease in the LCST. For all copolymers, a very sharp transition (when heated) was observed (see Fig. 3). A relatively small hysteresis (5 °C) on heating and cooling was observed, comparable with the hysteresis obtained for copolymers synthesized by ATRP.11
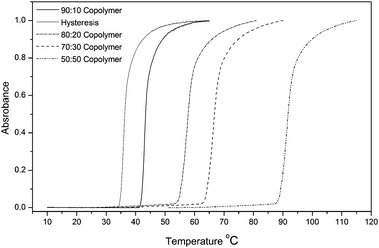 |
| | Fig. 3 UV-Vis plot of various copolymers to show the lower critical solution temperature (LCST) behavior in water. | |
Thiol–ene “click” of OEGMEMA oligomers
Catalytic chain transfer polymerization yields oligomers with terminal vinyl end-groups at high efficiency (almost 100%). This vinyl-functionalization is not possible to achieve by other common radical polymerization techniques, such as ATRP,56 NMP57 or RAFT.58 Since the introduction of the “click” concept by Sharpless et al.,27 there have been a number of efficient conjugation reactions designated as “click” reactions. Recently, Hawker et al.40 re-introduced an old but very efficient reaction, thiol–ene reactions, as a potential “click” reaction. Thiol–ene reactions embrace two types of mechanism: (i) anti-Markovnikov radical addition and (ii) base or nucleophile catalyzed Michael addition reaction.27,29,40,43,59–61 Thiol–ene reactions can be performed in different solvents and are tolerant of a large range of functionalities. Recent work has been performed on the nature of thiol–ene reaction conditions by Davis and Haddleton and Lowe and co-workers59 where it was reported that primary amines are more effective catalysts than secondary or tertiary amines, as well as significant reductions in the reactivity of methacrylates attributable to a combination of steric hindrance and inductive effects. The structure of the thiol compound also affects the efficiency of thiol–ene reactions, for example, alkyl thiols are less reactive than functional thiols, such as 2-mercaptoethanol or 1-thioglycerol. However, the previously reported studies were focused mainly on small molecules (monomers) or very short oligomers (dimers, trimers). In this current work it was necessary to optimize the thiol–ene Michael addition, as the “ene” components were methacrylate polymers to ensure high conversion and purity of the final product.
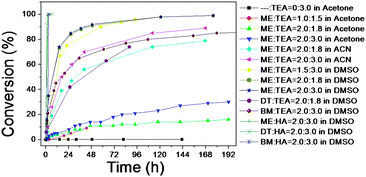
Poly(DEGMEMA) polymer was used to optimize the thiol–ene Michael addition of 2-mercaptoethanol. To establish the best solvent, the thiol–ene reactions were performed in a range of solvents: acetone, acetonitrile, and dimethyl sulfoxide (DMSO).
The following order of reactivity was established: DMSO > acetonitrile > acetone. The reaction carried out in DMSO reaches a very high yield at ambient temperature after 10 hours, while in acetone, yields superior to 30% could not be achieved. It is well known that polar solvents, such as DMSO, are good solvents for nucleophilic reactions.59b The efficacy of the catalyst used in these reactions, either phosphine or amine, was then examined. It has been highlighted previously that phosphine (e.g. dimethyl phenyl phosphine) is an effective catalyst30,59b at concentrations less than 0.05 mol% of the “ene” to prevent the side reaction of the conjugation of DMPP to the double bond.59a In this study, thiol–ene Michael reactions were initially performed and catalyzed by a combination of DMPP and TEA (the concentration of DMPP was maintained at less than 0.1 mol%), in which side reactions of DMPP conjugated to the vinyl end-group of poly(DEGMEMA) were observed (Fig. S2–S7, ESI†).
Subsequently, to minimize the formation of by-products, it was decided to use amine catalysts. Two different amines were compared: triethylene amine (TEA) (tertiary) and hexylamine (HA) (primary). The TEA catalyst ensured a full conversion of the vinyl end-group of poly(DEGMEMA), without the formation of by-products, when the reaction was performed with 2-mercaptoethanol at ambient temperature.59a
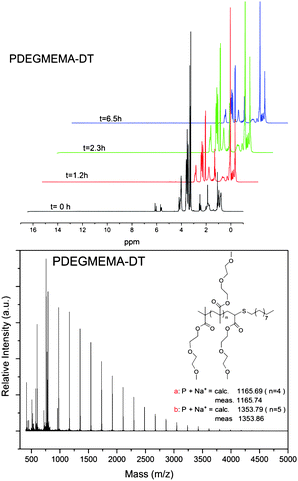
However, the efficiency of this TEA-catalyzed reaction drops when an aliphatic or aromatic thiol is used, such as benzyl mercaptan. (TEA can only drive the reaction to a maximum of 70% after 3 days with benzyl mercaptan at ambient temperature, Fig. 4.) Quantitative conversion could only be attained when the reaction was performed at 40 °C for 14 hours. In order to accelerate the rate and increase the conversion of the thiol-Michael addition reaction, HA was used as a catalyst.
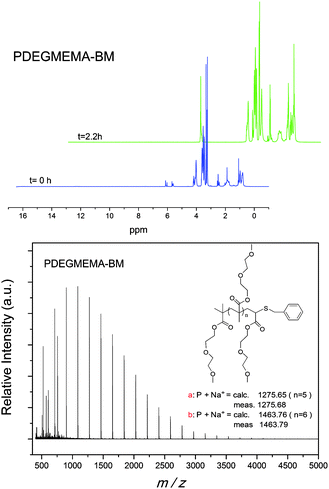
Thus, it was possible to functionalize different homopolymers of DEGMEMA and OEGMEMA475 with different functional thiols, such as benzyl mercaptan, 1-dodecanethiol, and 2-mercaptoethanol with a high efficiency. The detailed characterization of the “clicked” polymers was performed using 1H NMR, GPC as well as MALDI-ToF MS. The characterization data are presented in Fig. 5, 6 and 7 for 2-mercaptoethanol, 1-dodecanthiol and benzyl mercaptan, respectively.
Thermal analyses of post-functionalized poly(OEGMEMA)
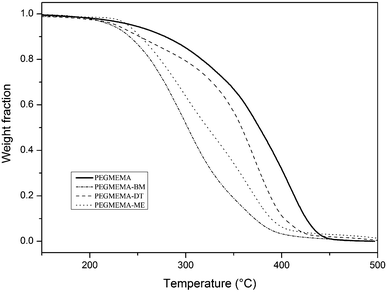
TGA and DSC of poly(OEGMEMA) were performed to show the influence of thiol–ene “click” products on the thermal properties of the polymers. The thermal gravimetry analysis data, as shown in Fig. 8, indicates that all thiol–ene modified poly(OEGMEMA) have lower thermal stability than poly(OEGMEMA) before modification (terminated by vinyl end-groups), regardless of the type of functional group. This result can be explained by the presence of the C–S bond, more unstable at high temperature than a C–C bond as indicated by the enthalpy of dissociation (Table 3).62,64
Table 3 Common bonds that can be found in the polymers with its corresponding enthalpy64
| Bond |
Enthalpy ΔH (kJ mol−1) at 25 °C |
| C–C |
346 |
| C–H |
414 |
| C–O |
358 |
| C–S |
289 |
| CC |
614 |
Further thiol–ene ‘click’ chemistry was performed on the copolymers of OEGMEMA475 and DEGMEMA to investigate the effect of different end-groups on their thermoresponsive behavior (LCST), Table 4. Recently Theato's group investigated copolymers made with various RAFT agents giving rise to a range of end-group functionalities and they investigated the influence of end-groups on LCST behavior.63 Homopolymers and copolymers functionalized by 1-dodecanethiol, benzyl mercaptan and 2-mercaptoethanol gave significantly different LCST temperatures attributable to hydrophobic and hydrophilic effects, Table 4. As expected, the thiol–ene “click” reaction of the hydrophobic end-group caused an LCST decrease while “click” reaction of the hydrophilic end-group lead the LCST to increase.
Table 4 List of polymers synthesized in this study using catalytic chain transfer polymerization and further modification by thiol-Michael addition to give an LCST librarya
| Run |
Structure |
M
n, GPC/g mol−1 |
PDI |
LCST/°C |
|
A2–A4 were obtained after thiol–ene modification with 2-mercaptoethanol, 1-dodecanethiol and benzyl mercaptan. A12 and A13 were obtained after thiol–ene modification with 1-dodecanethiol and 2-mercaptoethanol.
|
| A1 |
DEGMEMA |
2500 |
1.53 |
47.2 |
| A2 |
DEGMEMA–ME |
1100 |
2.91 |
52 |
| A3 |
DEGMEMA–DT |
3200 |
1.50 |
16 |
| A4 |
DEGMEMA–BM |
2200 |
1.61 |
32 |
| A5 |
PEGMEMA |
6000 |
1.40 |
>100 |
| CP1 |
DEGMEMA–PEGMEMA (50:50) |
2800 |
1.55 |
95 |
| CP2 |
DEGMEMA–PEGMEMA (70:30) |
2600 |
1.45 |
65 |
| CP3 |
DEGMEMA–PEGMEMA (80:20) |
2600 |
1.56 |
58 |
| CP4 |
DEGMEMA–PEGMEMA (90:10) |
3000 |
1.52 |
43 |
| A12 |
DEGMEMA–PEGMEMA (90:10)–DT |
3100 |
1.56 |
35 |
| A13 |
DEGMEMA–PEGMEMA (90:10)–ME |
3050 |
1.54 |
42 |
Conclusions
In conclusion, we report on the catalytic chain transfer polymerizations of oligo(ethylene glycol) methyl ether methacrylates using CoBF as a catalyst. The chain transfer values were measured to be similar with other (larger) methacrylates and also exhibit the same trends with the ester chain length. Moreover, copolymers of OEGMEMA475 and DEGMEMA have been synthesized and characterized in terms of thermal behavior and the lower critical solution temperature (LCST) in water. The terminal vinyl functionality of the oligomers has been exploited in thiol–ene reactions to make a range of functional oligomers.
Acknowledgements
CB and TPD thank ARC and UNSW for funding and for fellowship (CB and TPD: APD and Federation Fellow, respectively). DMH thanks CSC and UoW for funding (GL) and the EU MC fellowship (235999) (CRB). Equipment used in part was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF).
References
- A. A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753–1766 CrossRef CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611–3659 CrossRef CAS.
-
(a) J. P. A. Heuts, G. E. Roberts and J. D. Biasutti, Aust. J. Chem., 2002, 55, 381–398 CrossRef CAS;
(b) T. P. Davis, D. M. Haddleton and S. N. Richards, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1994, C34, 243–324 Search PubMed.
- T. P. Davis, D. Kukulj, D. M. Haddleton and D. R. Maloney, Trends Polym. Sci., 1995, 3, 365–373 CAS.
- J. P. A. Heuts, D. J. Forster and T. P. Davis, Macromolecules, 1999, 32, 3907–3912 CrossRef CAS.
- C. Kowollik and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3303–3312 CrossRef CAS.
- A. Debuigne, R. Poli, C. Jérôme and C. Detrembleur, Prog. Polym. Sci., 2009, 34, 211–239 CrossRef CAS.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378–2384 CrossRef CAS.
- M. E. Thomson, N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk and M. F. Cunningham, Macromolecules, 2010, 43, 5647–5658 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2005, 39, 893–896.
- C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef CAS; C. Boyer, M. R. Whittaker, K. Chuah, J. Liu and T. P. Davis, Langmuir, 2010, 26, 2721–2730 CrossRef CAS.
- S. A. Meenach, K. W. Anderson and J. Z. Hilt, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3229–3235 CrossRef CAS.
- M. Chanana, S. Jahn, R. Georgieva, J.-F. Lutz, H. Bäumler and D. Wang, Chem. Mater., 2009, 21, 1906–1914 CrossRef CAS.
- S. Kessel, S. Schmidt, R. Müller, E. Wischerhoff, A. Laschewsky, J.-F. Lutz, K. Uhlig, A. Lankenau, C. Duschl and A. Fery, Langmuir, 2009, 26, 3462–3467.
- H. M. Zareie, C. Boyer, V. Bulmus, E. Nateghi and T. P. Davis, ACS Nano, 2008, 2, 757–765 CrossRef CAS.
- E. Wischerhoff, S. Glatzel, K. Uhlig, A. Lankenau, J.-F. o. Lutz and A. Laschewsky, Langmuir, 2009, 25, 5949–5956 CrossRef CAS.
- E. Wischerhoff, N. Badi, J.-F. Lutz and A. Laschewsky, Soft Matter, 2010, 6, 705–713 RSC.
- Z. Zarafshani, T. Obata and J.-F. Lutz, Biomacromolecules, 2010, 11, 2130–2135 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138–7147 CrossRef CAS.
- T. M. Eggenhuisen, C. R. Becer, M. W. M. Fijten, R. Eckardt, R. Hoogenboom and U. S. Schubert, Macromolecules, 2008, 41, 5132–5140 CrossRef CAS.
- H. M. L. Thijs, C. R. Becer, C. Guerrero-Sanchez, D. Fournier, R. Hoogenboom and U. S. Schubert, J. Mater. Chem., 2007, 17, 4864–4871 RSC.
- C. Weber, C. R. Becer, W. Guenther, R. Hoogenboom and U. S. Schubert, Macromolecules, 2010, 43, 160–167 CrossRef CAS.
- C. Weber, C. R. Becer, R. Hoogenboom and U. S. Schubertt, Macromolecules, 2009, 42, 2965–2971 CrossRef CAS.
- C. R. Becer, K. Kokado, C. Weber, A. Can, Y. Chujo and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1278–1286 CrossRef CAS.
- H. Dong and K. Matyjaszewski, Macromolecules, 2010, 43, 4623–4628 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Polymer, 2009, 50, 3158–3168 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344–1350 CrossRef CAS.
-
(a) C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS;
(b) C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- G. Chen, S. Amajjahe and M. H. Stenzel, Chem. Commun., 2009, 1198–1200 RSC.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Sci., 2010, 20, 4745–4750 CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- M. W. Jones, G. Mantovani, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- N. Gupta, B. F. Lin, L. M. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V. Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138–145 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- F. A. Leibfarth, M. Kang, M. Ham, J. Kim, L. M. Campos, N. Gupta, B. Moon and C. J. Hawker, Nat. Chem., 2010, 2, 207–212 CrossRef CAS.
- A. Bakac, M. E. Brynildson and J. H. Espenson, Inorg. Chem., 1986, 25, 4108–4114 CrossRef CAS.
- A. Bakac and J. H. Espenson, J. Am. Chem. Soc., 1984, 106, 5197–5202 CrossRef CAS.
- A. A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1366–1376 CrossRef CAS.
- R. A. Sanayei and K. F. O'Driscoll, J. Macromol. Sci. Part A, 1989, 26, 1137–1149 CrossRef.
- T. P. Davis and D. Kukuly, Macromol. Chem. Phys., 1998, 199, 1697–1708 CAS.
- J. P. A. Heuts, L. M. Muratore and T. P. Davis, Macromol. Chem. Phys., 2000, 201, 2780–2788 CrossRef CAS.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378–2384 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- S. Harrison, F. Ercole and B. Muir, Polym. Chem., 2010, 1, 326–332 RSC.
- J. P. A. Heuts, D. Kukulj, D. J. Forster and T. P. Davis, Macromolecules, 1998, 31, 2894–2905 CrossRef CAS.
- J. F. Lutz and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 897–910 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS; C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
-
(a) G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC;
(b) J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
-
(a) C. Boyer, C. Loubat, J. J. Robin and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5146–5160 CrossRef CAS;
(b) C. Boyer, G. Boutevin, J. J. Robin and B. Boutevin, Polymer, 2004, 45, 7863–7876 CrossRef CAS;
(c) B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3235–3243 CrossRef CAS;
(d) C. Boyer, G. Boutevin, J.-J. Robin and B. Boutevin, Macromol. Chem. Phys., 2004, 205, 645–655 CrossRef CAS.
- J. D. Coyle, Tetrahedron, 1985, 41, 5393–5425 CrossRef CAS.
- P. J. Roth, F. D. Jochum, F. R. Forst, R. Zentel and P. Theato, Macromolecules, 2010, 43, 4638–4645 CrossRef CAS.
-
G. Aylward and T. Findlay, SI Chemical Data, John Wiley & Sons, Milton, QLD, 5th edn, 2002, p. 121 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.