Hybrid biological spores wrapped in a mesh composed of interpenetrating polymer nanoparticles as “patchy” Pickering stabilizers†
Nicholas
Ballard
and
Stefan A. F.
Bon
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: s.bon@warwick.ac.uk; Web: www.stefanbon.eu
First published on 2nd December 2010
Abstract
We describe a new method for the decoration of the intricate morphology of spore particles with polymer nanoparticles and investigate their behaviour at liquid–liquid interfaces. We found a large difference in the interfacial activity between spherical microspheres and the anisotropic particles synthesized here and describe this in terms of particle wettability.
Introduction
Nature shows a fascinating complexity in the morphological structure of small objects. Unicellular organisms such as diatoms are provided with a captivating nanopatterned biomineralized armor1 whilst pollen grains and spores show intricate variety in shape, patterns, and chemical composition.2 The anisotropy in structure generates advanced functionalities, for example optimization of adhesion and transport characteristics. Drawing inspiration from this a revolution in the fabrication of manmade anisotropic colloidal particles is taking place, as variety in shape and chemical or morphological patchiness offers great engineering potential in advanced supracolloidal material design. Particles of amphiphilic Janus-type nature, for example, have the ability to self-assemble into suprastructures analogous to those formed by amphiphilic molecules,3 the assembly process driven by hydrophobic attractive forces and governed by a geometric packing parameter. Noticeable commercialized examples are in the area of electronic paper and e-books, using bichromal dipolar microspheres imbedded in a soft fluidic matrix as tech-base,4 or exploiting electrophoretic display technology in order to dynamically induce anisotropy in microcapsule content.5 Numerous innovative techniques have been developed in order to produce anisotropic particles with wide-ranging morphologies from ellipsoids6–8 and cylinders9,10 to dumbbell-like particles.11–13 Despite these advances synthetic fabrication efforts still pale into insignificance in terms of the complexity and scale of production when compared to microscopic particles made by nature. A promising route towards complex anisotropic colloids is to transform naturally occurring particles into synthetic hybrids. Biological small objects are used as a template so that morphological complexity is preserved or replicated. In addition functionality can be tailored through chemical modification. Mann and coworkers used a variety of pollen grains as templates to fabricate inorganic replicas made from SiO2, CaCO3, and calcium phosphates by soaking the structures in a precursor solution which were later precipitated.14 More complex structures with functional properties have been replicated. For example, the intricate structures of butterfly wings were used as a photonic scaffold for controlled assembly of CdS nanoparticles by activating the original wing to serve as a reactive site for the deposition of the inorganic nanocrystallites, producing materials that exhibited interesting optical properties.15Instead of synthesizing inorganic replicas of biological objects herein we show that we can selectively coat Lycopodium clavatum spores with crosslinked polymer nanoparticles through a free radical precipitation polymerization process. The in situ formed polymer colloids self-assemble on specific parts of the spore surface forming an interpenetrating polymer mesh. We can tailor the fabrication of the “patchy” hybrid biological spores with control over the thickness of the mesh as well as its chemical composition and thus functionality. We investigated the ability of these “patchy” hybrid spores to adhere to liquid–liquid interfaces and serve as Pickering stabilizers. Their adhesion behaviour could be influenced upon variation of the chemical composition, and thus lyophilicity, of the polymer mesh. We show that the anisotropic polygon-like “patchy” morphology of the hybrid spores substantiates a marked and distinct wettability in comparison to isotropic microspheres.
Lycopodium spores are commercially available in large quantities and have frequently proved to be useful throughout history in applications ranging widely from herbal remedies to pyrotechnics.16 These spores, as is common to many other pollinating particles, are of complex morphology and monomodal in size. The 30 µm Lycopodium particles used here have a unique structure with a polygon-like patterned surface consisting of high ridges. The chemical structure of the spores is rather ill-defined but can be separated into two distinct regions. The inner layer of the spore wall is composed of polysaccharides whereas the external ridge-like layer of the particle is made up of sporopollenin, an extremely resistant biopolymer derived from the oxidative polymerization of carotenoids and carotenoid esters.17
Results and discussion
We chose to use free radical precipitation polymerization of divinylbenzene (main monomer) as the method by which to selectively coat the spores with functional polymeric particles (see Fig. 1). Precipitation polymerization of divinylbenzene (DVB), commonly carried out in acetonitrile, is an established method for the synthesis of monodisperse, heavily crosslinked, micron-sized particles with extensive amounts of work on core–shell and copolymer particles.18–20 The growth mechanism of the particles throughout the reaction has been discussed in detail by Stöver and coworkers.21 The initial homogeneous mixture of solvent, monomer(s), and initiator is heated so that oligomeric radicals form in solution. Further propagation occurs in solution up to the point at which the fractal-like chains reach a critical molar mass upon which they aggregate and desolvate to form the particle nuclei. The formed nuclei capture oligomeric chains from solution throughout the polymerization and grow continuously as the reaction proceeds.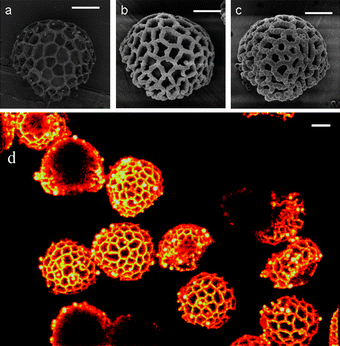 | ||
| Fig. 1 Selective adsorption of polymer particles to the surface of Lycopodium spores at various points of the reaction. (a) 0 hours, (b) 8 hours and (c) 24 hours. The scale bar in all cases is 10 µm. (d) Confocal laser scanning image Z stack projection (64 z slices of 0.4 µm) of particles after DVB copolymerization at 70 °C for 24 hours with fluorescent tag hostasol methacrylate. | ||
In our experiments the presence of the micron-sized Lycopodium spores induces heterocoagulation of the polymer particles onto the surface of the spores resulting in complex polymer architectures (see Fig. 1 and ESI†). In all cases the polymer only adsorbed onto the outer polygon-like surface of the spore particles thereby forming an interpenetrating mesh. This was visualised with greater ease by confocal microscopy on hybrid Lycopodium particles that were fabricated through copolymerizing a small amount of fluorescent monomer, in this case a hostasol functional methacrylate22 (see Fig. 1d). The ridges of the spore particles are saturated but there is no evidence of adsorption inside the holes between the ridges. It is thought that this spatially selective heterocoagulation is the result of a long-range hydrophobic interaction between the spore ridge, composed of sporopollenin, and the emerging polymer particle mesh. Subsequent growth of the interpenetrating polymer mesh occurs as the oligomeric chains in solution are captured.
A series of precipitation polymerizations of divinylbenzene (5.0 vol%) was performed in acetonitrile in the presence of azobisisobutyronitrile (AIBN) as thermal radical initiator (3 wt% to DVB). The weight ratio of Lycopodium spores to monomer was varied (L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M = 0, 0.5, 1.0 and 2.0 respectively) in order to investigate polymerization kinetics and the growth of the interpenetrating polymer mesh. Precipitation polymerizations of DVB carried out in the absence of Lycopodium were colloidally unstable and coagulated after approximately 3 hours. Monomer conversion, XM, monitored by 1H NMR comparing the vinyl hydrogens of divinylbenzene to a known amount of ethyl acetate tracer, indicated the polymerization was first order with respect to divinylbenzene when initiator decomposition was taken into account in all cases. This result is expected since propagation mainly occurs in solution so follows the kinetics of a typical solution polymerization. The kinetic plots with varying L:M are available as the ESI†.
:M = 0, 0.5, 1.0 and 2.0 respectively) in order to investigate polymerization kinetics and the growth of the interpenetrating polymer mesh. Precipitation polymerizations of DVB carried out in the absence of Lycopodium were colloidally unstable and coagulated after approximately 3 hours. Monomer conversion, XM, monitored by 1H NMR comparing the vinyl hydrogens of divinylbenzene to a known amount of ethyl acetate tracer, indicated the polymerization was first order with respect to divinylbenzene when initiator decomposition was taken into account in all cases. This result is expected since propagation mainly occurs in solution so follows the kinetics of a typical solution polymerization. The kinetic plots with varying L:M are available as the ESI†.
The formation and growth of the polymer mesh composed of interpenetrating polymer nanoparticles onto the surface of the Lycopodium spores was monitored by SEM as a function of monomer conversion and expressed in approximate thickness. Experiments at a L:M of 0.5 showed rapid saturation of the ridges of the Lycopodium spores with interpenetrating polymer nanoparticles. Secondary homogeneous nucleation occurred after 2 hours, leading to the formation of a crop of dispersed spherical micron-sized DVB particles, as detected by an increase in turbidity. Higher concentrations of the Lycopodium particles led to a slower increase in thickness of the polymer mesh, with secondary nucleation in the continuous phase completely suppressed for L:M = 2.0. In this case the larger surface area facilitated a greater volume of polymer to precipitate onto the ridges thus preventing secondary nucleation.
The growth (thickness) of the polymer mesh on the surface of the Lycopodium spores can easily be modelled when we ignore secondary homogeneous nucleation. Expressing the polymer mesh as a continuous non-self-avoiding cylinder with radius R and length l, and estimating the total polymer mesh volume, V, by summation over the number of particles, Nspore, we can correlate monomer conversion as follows:
| XM ∝ V ∝ πR2Nsporel | (1) |
N
spore is calculated using a density value of 0.34 g cm−3 for the Lycopodium spores. An approximate value for l, the total length of the ridged surface of one spore, of 750 µm was obtained from SEM analysis of a batch of Lycopodium spores. In the absence of secondary nucleation a graph of XM plotted against R2Nspore should yield a straight line with gradient πl (see Fig. 2). It can be seen that for low L:M the plot deviates significantly from the theoretical line, where R2Nspore is calculated by dividing the volume of polymer at a given time (calculated from kinetic experiments) by πl (l = 750 µm), indicating secondary nucleation. For the case of high values of L:M where little or no individual polymer particles were observed we see good agreement with the theoretical line.
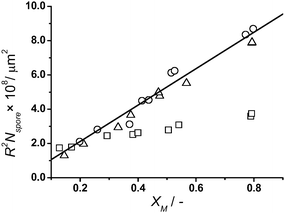 | ||
| Fig. 2
Growth of polymer on spore particle modelled as a continuous cylinder at varying L:M values: □ L:M = 0.5, △ L:M = 1, and ○ L:M = 2. The straight line is the theoretical increase of R2Nspore with conversion calculated using l = 750 µm. | ||
Interfacial activity of patchy particles
The chemical composition of the polymer mesh wrapped around the Lycopodium spores could be readily altered upon copolymerization with functional monomers, in our case methacrylic acid (MAA), 2-hydroxyethyl methacrylate (HEMA), and methyl methacrylate (MMA) all in a 1:1 weight ratio with respect to DVB. Polymerizations were stopped after 5 h in order to restrict secondary nucleation, using L:M = 1.0. The hybrid spores were subsequently cleaned by repeated sedimentation and washing with acetone followed by drying in a vacuum oven at 40 °C. The prime reason to vary chemical composition was to influence the leophilicity of the hybrids, in other words their affinity to wet either water or oil (hydrophilic versus hydrophobic). Control of this would allow fine-tuning of their adhesion properties to soft interfaces, and thus their efficacy as Pickering stabilizers. Binks et al.23 already reported that pristine Lycopodium spores can stabilize various oil-in-water emulsions. Our interest was to explore the role of the “patchy” polygon-like morphology of the spores. As a model system we examined the use of our hybrid “patchy” particles to stabilize emulsions of hexadecane and water. The particles were suspended in hexadecane (4 wt%) and mixed with an equal volume fraction of water upon which emulsions were formed by handshaking and left for 1 hour. To our initial surprise the most hydrophobic hybrid particles, where the polymer only consisted of divinylbenzene, were unable to emulsify any oil. In sharp contrast the two components of the structured particles, the Lycopodium spore and a DVB sphere, are both excellent Pickering stabilizers. The wettability of the particles, which directly correlates to the interfacial adsorption activity, can therefore be described as a function of both the surface chemistry and the surface roughness.24,25 A polymer mesh composed of DVB and MMA to lessen its hydrophobicity only led to negligible improvements. Copolymerization with more hydrophilic monomers, that is MAA and HEMA, led to stable oil-in-water emulsions that remained stable for a period of many weeks. The wettability of the hybrid spores and hence their capability to serve as Pickering stabilizers were further investigated by determination of their apparent three phase contact angle, θ, using the “gel trapping” technique (see Fig. 3).26Lycopodium spores wrapped in a mesh of poly(DVB) did not wet the oil–water interface at all (image omitted). Upon incorporation of methyl methacrylate, a more hydrophilic monomer, the spores exhibited limited adhesion to the oil–water interface (θ ≈ 10°, Fig. 3b). When the hydrophilic co-monomers, HEMA and MAA, were used the contact angle was closer to 90° (θ ≈ 105° and θ ≈ 130° respectively), giving the emulsion increased stability.
 | ||
| Fig. 3 SEM image of the Lycopodium spores position at the oil–water interface for (a) bare Lycopodium, (b) pDVB-co-pMMA coated Lycopodium, (c) pDVB-co-pHEMA coated Lycopodium and (d) pDVB-co-pMAA coated Lycopodium. Scale bar in all cases is 10 µm. | ||
The results found experimentally agree with theoretical determination of the position of the spore at the interface. The position of a particle at a heterogeneous interface has traditionally been described by computing the free energy change based on interfacial tension as originally demonstrated by Pieranski.27 The surface activity of non-uniform particles has recently been investigated in detail by Nonomuraet al.28–30 who evaluated the interfacial energy as a function of a roughness factor. Using a similar approach we model the spore particles as a uniform Archimedean solid. For a non-spherical particle the interfacial energy, ET, is the sum of the energy of the particle–oil interface, EPO, the particle–water interface, EPW, and the missing oil–water interface, EOW.
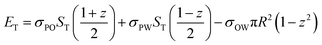 | (2) |
Knowing the value of ST for the structure of interest we can use eqn (2) to compute the free energy of the system through the liquid–liquid interface. The energy minima, obtained by differentiation of eqn (2), define the vertical coordinate at equilibrium.
 | (3) |
This allows us to explain for the observation that a sphere of poly(DVB) and a spore particle are adsorbed at the liquid–liquid interface but our hybrid spores of DVB, where both the interfacial tension term and the total surface area are large, are not. For the case of a polystyrene sphere, which has similar interfacial tension to DVB, the energy minima are found at z = 0.44 corresponding to a contact angle of approximately 60° whereas the corresponding buckyball type structure with identical interfacial tension values but an increased surface area has an energy minima at z = 0.9 with contact angle approximately 30° which results in significantly reduced interfacial activity. This is shown graphically in Fig. 4. Similarly Fig. 4 shows that for a more hydrophilic poly(HEMA) coated spore we expect a shift in contact angle to reflect the increase in surface area from the case of a sphere to a buckyball type structure. These theoretical results can be used to explain the experimentally determined position of the copolymer hybrid spores at the interface shown in Fig. 3. For polymer particles which are known to adsorb at interfaces we see their position at the interface accentuated towards either the oil phase, in the case of MMA and to the point of complete wetting for DVB, or towards the aqueous phase, for copolymers of HEMA and MAA due to the large increase in surface area.
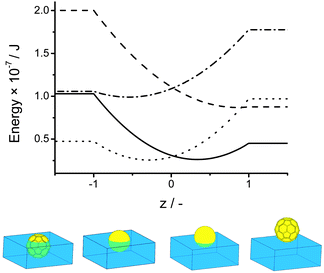 | ||
Fig. 4 Position of a sphere of polystyrene (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and of polyHEMA (⋯) and a buckyball type structure of polystyrene ( ) and of polyHEMA (⋯) and a buckyball type structure of polystyrene (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and polyHEMA ( ) and polyHEMA (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ) at the oil–water interface interface and illustration of predicted contact angles for (from left to right) a buckyball type structure of polyHEMA, a polyHEMA sphere, a polystyrene sphere and a polystyrene buckyball structure. ) at the oil–water interface interface and illustration of predicted contact angles for (from left to right) a buckyball type structure of polyHEMA, a polyHEMA sphere, a polystyrene sphere and a polystyrene buckyball structure. | ||
Conclusions
We have developed an original method for selective coating of micrometre sized naturally occurring spore particles with smaller sub-micrometre particles via an in situprecipitation polymerization. By fine tuning the monomer mixture the surface properties of the spore particles can be easily altered and this has been demonstrated by use of the gel trapping technique to exert control over the three phase contact angle at the oil–water interface demonstrating that shape anisotropy has a huge impact on the stability of particle stabilized emulsion droplets.Experimental
Materials
Divinylbenzene (DVB, 80%, mixture of isomers), 2-hydroxyethyl methacrylate (HEMA, 98%) and methyl methacrylate (MMA, >99%) were purchased from Sigma-Aldrich and were filtered through a short column of basic alumina before use to remove inhibitor. Lycopodium powder and phytagel were purchased from Sigma-Aldrich and were used without further purification. Azobisisobutyronitrile (AIBN) was purchased from Wako and used without further purification. All solvents were of reagent grade purity and were purchased from Fisher scientific. Sylgard 184 was purchased from Farnell UK and used in a ratio of 9:1 with respect to curing agent.
Equipment
Scanning electron microscopy was performed on a Zeiss supra 55VP FEGSEM. Particles were coated with a thin layer of platinum prior to scanning. Confocal imaging was performed on a Zeiss LSM 510 confocal microscope. NMR measurements were performed on a Bruker DPX-300 300 MHz spectrometer and the spectrum was analysed with Mestrec v2.3a.Synthesis
In a typical experiment AIBN (10 mg, 3 wt% wrt monomer), the monomer(s) (300 mg) and Lycopodium spores (300 mg) were dissolved in acetonitrile (15 ml), degassed and sealed in a vial with screw cap lid. The mixture was placed in an oven with a rotating steel plate at 25 rpm and heated slowly over the course of 1 hour to 70 °C. The mixture was then left for 5 hours before being removed and left to cool. The particles were left to settle then the supernatant liquid was decanted and the particles resuspended in acetone and the process repeated 5 times. The particles were then left to dry in air.Pickering emulsions
Particle stabilized emulsions were generated by mechanical shaking of a hexadecane water mixture. The spore particles were dispersed in hexadecane due to their hydrophobicity at a weight fraction of up to 4% and water was added (usually at a 1:1 ratio with respect to the oil phase). The vial was attached to a mechanical shaker for 10 minutes or alternatively shaken by hand for a similar amount of time. The emulsion type was inferred by placing a small amount of dye in the aqueous phase.
Acknowledgements
Equipment used was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). We thank Unilever for financial support (NB).References
- F. E. Round, R. M. Crawford and D. G. Mann, The Diatoms. Biology and Morphology of the Genera, Cambridge University Press, 1990 Search PubMed.
- J. Heslop-Harrison, Science, 1968, 161, 230–237 CrossRef CAS.
- S. Whitelam and S. A. F. Bon, J. Chem. Phys., 2010, 132, 074901 CrossRef.
- T. Nisisako, T. Torii, T. Takahashi and Y. Takizawa, Adv. Mater., 2006, 18, 1152–1156 CrossRef CAS.
- Y. Chen, J. Au, P. Kazlas, A. Ritenour, H. Gates and M. McCreary, Nature, 2003, 423, 136–136 CrossRef CAS.
- J. A. Champion, Y. K. Katare and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11901–11904 CrossRef CAS.
- P. Jiang, J. F. Bertone and V. L. Colvin, Science, 2001, 291, 453–457 CrossRef CAS.
- C. C. Ho, A. Keller, J. A. Odell and R. H. Ottewill, Colloid Polym. Sci., 1993, 271, 469–479 CrossRef CAS.
- S. Badaire, C. Cottin-Bizonne, J. W. Woody, A. Yang and A. D. Stroock, J. Am. Chem. Soc., 2007, 129, 40–41 CrossRef CAS.
- C. J. Hernandez and T. G. Mason, J. Phys. Chem. C, 2007, 111, 4477–4480 CrossRef CAS.
- H. R. Sheu, M. S. Elaasser and J. W. Vanderhoff, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 629–651 CrossRef CAS.
- J. W. Kim, R. J. Larsen and D. A. Weitz, Adv. Mater., 2007, 19, 2005–2009 CrossRef CAS.
- S. M. Yang, S. H. Kim, J. M. Lim and G. R. Yi, J. Mater. Chem., 2008, 18, 2177–2190 RSC.
- S. R. Hall, H. Bolger and S. Mann, Chem. Commun., 2003, 2784–2785 RSC.
- J. Han, H. L. Su, D. Zhang, J. J. Chen and Z. X. Chen, J. Mater. Chem., 2009, 19, 8741–8746 RSC.
- E. S. Rajendran, Homeopathy, 2004, 93, 99–102 CrossRef CAS.
- J. Brooks and G. Shaw, Nature, 1968, 219, 532–533 CrossRef CAS.
- W. H. Li and H. D. H. Stover, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2899–2907 CrossRef CAS.
- W. H. Li and H. D. H. Stover, Macromolecules, 2000, 33, 4354–4360 CrossRef CAS.
- W. H. Li, K. Li and H. D. H. Stover, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2295–2303 CrossRef CAS.
- J. S. Downey, R. S. Frank, W. H. Li and H. D. H. Stöver, Macromolecules, 1999, 32, 2838–2844 CrossRef CAS.
- F. Tronc, M. Li, J. P. Lu, M. A. Winnik, B. L. Kaul and J. C. Graciet, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 766–778 CrossRef CAS.
- B. P. Binks, J. H. Clint, G. Mackenzie, C. Simcock and C. P. Whitby, Langmuir, 2005, 21, 8161–8167 CrossRef CAS.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 8622–8631 CrossRef CAS.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2001, 17, 4540–4547 CrossRef CAS.
- V. Paunov, Langmuir, 2003, 19, 7970–7976 CrossRef CAS.
- P. Pieranski, Phys. Rev. Lett., 1980, 45, 569–572 CrossRef CAS.
- Y. Nonomura, S. Komura and K. Tsujii, J. Phys. Chem. B, 2006, 110, 13124–13129 CrossRef CAS.
- Y. Nonomura and S. Komura, J. Colloid Interface Sci., 2008, 317, 501–506 CrossRef CAS.
- Y. Nonomura, S. Komura and K. Tsujii, Langmuir, 2005, 21, 9409–9411 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental detail, high magnification SEM images, and kinetic data are included. See DOI: 10.1039/c0py00335b |
| This journal is © The Royal Society of Chemistry 2011 |