Poly(N-isopropylacrylamide) branched polymer nanoparticles
Pierre
Chambon
a,
Lin
Chen
a,
Steve
Furzeland
b,
Derek
Atkins
b,
Jonathan V. M.
Weaver
*c and
Dave J.
Adams
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: d.j.adams@liverpool.ac.uk
bUnilever R&D, Colworth Science Park, Sharnbrook, Bedford, MK44 1LQ, UK
cDepartments of Materials and Bioengineering, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: j.weaver@imperial.ac.uk
First published on 24th January 2011
Abstract
A series of thermally responsive branched copolymers based on N-isopropylacrylamide (NIPAAm) and poly(ethylene glycol) methacrylate (PEGMA) are synthesized using a direct, single step modified free radical polymerization strategy. The aqueous solution behaviour and nanoparticle formation of these copolymers are studied and correlated to the NIPAAm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEGMA molar ratio and PEGMA chain length. All copolymers are water-soluble as discrete polymer chains at low temperature. On raising the solution temperature, inter-polymer interactions—that is, aggregation of polymers—are inhibited at higher PEGMA molar ratios and at long PEGMA chain lengths. The statistical incorporation of monomer residues and the short primary chain lengths within these branched copolymer structures result in the effective “dampening out” of NIPAAm thermo-responsivity and provides new insight into the architecture and composition of these complex copolymer structures.
:PEGMA molar ratio and PEGMA chain length. All copolymers are water-soluble as discrete polymer chains at low temperature. On raising the solution temperature, inter-polymer interactions—that is, aggregation of polymers—are inhibited at higher PEGMA molar ratios and at long PEGMA chain lengths. The statistical incorporation of monomer residues and the short primary chain lengths within these branched copolymer structures result in the effective “dampening out” of NIPAAm thermo-responsivity and provides new insight into the architecture and composition of these complex copolymer structures.
Introduction
Thermoresponsive polymers have attracted considerable attention,1 with poly(N-isopropylacrylamide) (pNIPAAm) being perhaps the most studied. pNIPAAm is soluble in water below its lower critical solution temperature (LCST), but undergoes phase separation at higher temperature as the hydrogen bonds with water that solubilise the polymer break down. There are a large number of examples examining the aggregation of pNIPAAm-based block copolymers. These include block copolymers where the pNIPAAm block acts as the hydrophobic source of a double-hydrophilic system such as poly(ethylene oxide)-b-pNIPAAm2–7 or pNIPAAm grafted with PEG blocks.8–12 The aggregation and self-assembly of PEO-b-pNIPAAm copolymers in aqueous solution with temperature has been studied extensively over the last few decades, and the effect of the polymer architecture has been keenly studied. In general, the copolymers are soluble in water below the critical aggregation/micellisation temperature (CAT or CMT), which is usually close to the LCST of pNIPAAm, and form core–shell micellar aggregates at elevated temperatures. Modifying linear pNIPAAm with short PEO grafts produced polymers with a single transition in aqueous solution in which the chains collapse to form core–shell nanostructures.12 Later work showed that the PEO content strongly affects the temperature of this phase transition.10,11 Recent studies have shown evidence for association at low temperature due to the incompatibility between PEO and pNIPAAm.4 Thus the parameters influencing their self-assembly and final micellar size are well understood and include the ratio of PEO to pNIPAAm, the architecture of the polymers, the polymer concentration, salts and the heating rate.2,5,7,9,13,14Different PEO–pNIPAAm architectures have been examined including linear block copolymers, random copolymers, star polymers and graft copolymers. Recently, interest has been gathering in branched polymers as a means of generating more complex polymer architectures.15–19 In 2000, Sherrington and co-workers reported a generic and scalable thiol regulated free-radical polymerization process, which allows the production of branched vinyl polymers.20–22 This technique uses bifunctional monomers such as ethylene glycol dimethacrylate as branching points in a conventional free-radical polymerization. Polymer gelation is avoided by adding a chain transfer agent (CTA).20–24 This results in a reduction in the primary chain length to below the value that results in gelation. The degree of branching can be varied by altering the molar ratio of the monofunctional monomer relative to the bifunctional monomers. At higher degrees of branching, higher CTA concentrations (or CTAs with higher chain transfer coefficients) are required to inhibit gelation. One of us reported the synthesis of branched pH-responsive copolymers by the CTA mediated branched copolymerisation of poly(ethylene glycol) methacrylate (PEGMA) and N,N-diethylaminoethyl methacrylate (DEAEMA).25 These polymers were found to have similarities to micellar structures formed by the aggregation and subsequent cross-linking of PEO-b-pDEAEMA-based copolymers, so-called shell cross-linked micelles.15 At basic pH, the copolymers form reasonably well-defined micellar structures whose hydrodynamic diameters vary in diameter between 16 and 46 nm depending on the degree of branching. On reducing the solution pH, the branched copolymers were hydrated and swell similarly to pH-responsive shell cross-linked micelles.26 Thus this polymerization procedure represents a convenient and potentially scalable one-step strategy to polymeric (responsive) nanoparticles without recourse to time consuming self-assembly and cross-linking steps.
Here, we use this branching technology to prepare a range of thermally responsive branched copolymers based on PEGMA and NIPAAm. The structural and compositional diversity afforded by this approach provides a viable method to probe the effect of the PEGMA chain length and molar fraction on the thermally induced association (and thus nanoparticle formation) behaviour of this new class of copolymer.
Experimental
Materials
Ethylene glycol dimethacrylate (EGDMA) was purchased from Aldrich and passed through an alumina column prior to use in order to remove inhibitors. Poly(ethylene glycol) methacrylate (Mn = 2080 g mol−1) (PEGMA(2K)), 50 wt% in water, was purchased from Aldrich and freeze-dried prior to use. N-Isopropylacrylamide (NIPAAm), poly(ethylene glycol) methacrylate (Mn = 1100 g mol−1) (PEGMA(1K)), 1-dodecanethiol (DDT) and α,α′-azobisisobutyronitrile (AIBN) in solution in toluene (0.2 M) were obtained from Aldrich and used as received. Toluene (Fisher Scientific, analytical reagent grade) was degassed by bubbling nitrogen through for 30 minutes prior to use. The molecular weights of the quoted poly(ethylene glycol) methacrylates are those provided by the supplier.Preparation of branched polymers
NIPAAm, PEGMA (1K or 2K), EGDMA and the Chain Transfer Agent (dodecanethiol, DDT) were introduced in the desired molar ratio into a two-necked round bottom flask equipped with a stirrer bar and purged with a flow of nitrogen for 15 min. Degassed toluene (10 w/v% based on the total monomer) was added to the mixture of reactants and heated to 70 °C. Once the required temperature had been reached, the polymerisation was started by addition of AIBN (1.5% based on the total number of moles of double bonds) and left stirring for 48 hours. After this time, the toluene was removed by evaporation at reduced pressure. An aliquot of crude product was taken for further analysis. The crude product was then dissolved in a minimum amount of CHCl3 and precipitated into cold diethyl ether. The resulting product was dried overnight in a vacuum oven.In a typical synthetic procedure to prepare PEGMA(1K)5/NIPAM95–EGDMA15–DDT15 branched copolymer, PEGMA(1K) (1.000 g, 0.91 mmol), NIPAAm (1.955 g, 17.29 mmol), EGDMA (0.541 g, 2.73 mmol), DDT (0.552 g, 2.73 mmol), toluene (40 mL) and AIBN (58 mg, 0.35 mmol) were introduced in a reactor according to the procedure described above. After 48 hours at 70 °C, the polymer was collected and purified by precipitation into diethyl ether.
Characterisation
Results and discussion
Polymer synthesis
The branched copolymers were prepared using NIPAAm and PEGMA, with EGDMA as branching agent and 1-dodecanethiol (DDT) as the chain transfer agent. A typical synthetic procedure is shown in Scheme 1. Depending on the polymer to be prepared, the molar ratios of PEGMA:NIPAAm were either 5:95 or 15:85 and the molecular weights of the PEGMA macromonomers used were either 1100 g mol−1 or 2080 g mol−1 (see Table 1). The amount of CTA and EGDMA required to form a branched copolymer but prevent gelation has been previously discussed.25 Therefore, as described elsewhere, we used stoichiometric ratio of EGDMA:CTA. By way of an example, we refer to branched copolymers synthesized with the feed molar ratios of PEGMA–NIPAAm–EGDMA–DDT of 5:95:15:15 and PEGMA whose molecular weight is 1100 g mol−1 as PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15. The reactivity ratios for NIPAAm and oligo(ethylene glycol) methacrylates have been measured and show that the methacrylate has a higher reactivity.27 This implies that, at low NIPAAm conversion, the polymers will be relatively rich in PEGMA, with NIPAAm-rich polymer chains (relative to the initial feed ratio) being formed at a later stage of the copolymerization reaction. However, the polymer chains are conjoined and this statistical process will ‘correct’ the compositional drift to some degree. A range of branched copolymers with varying ratios of PEGMA–NIPAAm and molecular weight of PEGMA were synthesized, as well as a linear copolymer obtained under the same conditions in the absence of EGDMA (cf.Table 1, entry 5). In all cases, the monomer conversions (based on alkenic protons by NMR) were greater than 95%, although accurate determination of conversion was difficult due to the overlapping NH and alkene resonances.
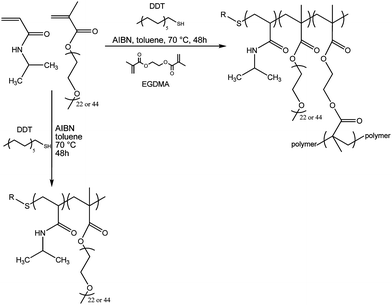 | ||
| Scheme 1 Schematic representation of the synthesis of the PEGMA–NIPAAm based branched and linear copolymers. | ||
| Target polymer composition | PEGMA molecular weight/g mol−1 | EGDMA a | Polymer compositionb | Yieldc | M n/g mol−1 | M w/g mol−1 | M w/Mn | α |
|---|---|---|---|---|---|---|---|---|
| a Target molar equivalent based on the monofunctional monomer nominally set to100. b Polymer composition calculated by 1H NMR excluding the CTA and EGDMA due to overlapping proton resonances. c Gravimetric yields after purification by precipitation. d Determined by GPC in THF. e Determined by GPC in DMF. f Determined by GPC in THF by triple detection methods. g Crude product. | ||||||||
| PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 | 1100 | 15 | PEGMA4/NIPAAm96 | 54% | 5100d, 8400e, 9300f | 7200d, 12700e, 15000f |
1.4d, 1.5e, 1.6f | 0.32f |
| PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15 | 2080 | 15 | PEGMA8/NIPAAm92 | 80% | 5600d, 7700e, 6400f | 8150d, 12200e, 11400f |
1.5d, 1.6e, 1.8f | 0.21f |
| PEGMA(1K)15/NIPAAm85–EGDMA15–DDT15 | 1100 | 15 | PEGMA14/NIPAAm86 | 74% | 6300d, 6300e, 8400f | 7300d, 8300e, 14700f |
1.2d, 1.3e, 1.8f | 0.29f |
| PEGMA(2K)15/NIPAM85–EGDMA15–DDT15 | 2080 | 15 | PEGMA15/NIPAAm85 | 59% | 6500d, 11100e, 6400f |
7700d, 16000e, 12200f |
1.2d, 1.4e, 1.9f | 0.26f |
| PEGMA(1K)5/NIPAAm95–DDT15 | 1100 | 0 | —g | 1200d | 1800d | 1.5d |
The molar ratios of PEGMA and NIPAAm in the purified branched copolymers were estimated by 1H NMR in CDCl3 (Fig. 3). Despite the majority of the representative peaks overlapping, a reasonable assessment of the molar ratio incorporation of PEGMA and NIPAAm is possible by considering the three protons of the PEGMA chain end (–O–CH3, δ 3.3 ppm) and the six protons of the pNIPAAm (–NH–CH–(CH3)2, δ 1.1 ppm). These estimated values are gathered in Table 1 and are in good agreement with those targeted. Unfortunately, it was not possible to assess the amount of EGDMA and CTA due to overlapping and broad signals.
The molecular weights were initially determined by GPC in THF at 40 °C. All the samples were completely soluble in THF and exhibited a relatively low molecular weight (below 10000 g mol−1), Fig. 1. We have previously discussed the difficulties in analysing similar branched polymers by GPC—including triple detection GPC.15,28 The GPC data recorded for such complex architecture polymers are extremely complicated and we do not believe that the recorded molecular weight parameters are necessarily absolute and favour the use of systematic trends between small polymer libraries as a descriptor of molecular weight characteristics. Systematic GPC analyses were performed on these polymer systems using both triple detection GPC and single detection methods with different columns. The most reproducible trends were found using the single detection methods. This methodology, however, may account for the low molecular weights determined in comparison to other work using triple detection methods. The GPC chromatograms of the branched copolymers displayed relatively narrow polydispersities. Indeed, the PEGMA rich branched copolymers displayed polydispersities around 1.2, which is similar to values obtained by controlled living polymerisation. These observations are similar to those found previously for similar branched polymers prepared using PEGMA and DEAMA with DDT as the CTA.25 It is noteworthy that the GPC traces of the branched copolymers synthesised here are significantly separated from the trace of the linear sample made in the same condition (Fig. 1). Moreover, the molecular weights of the branched copolymers are more than three times higher than the one of the linear sample, showing the effective incorporation of the EGDMA brancher.
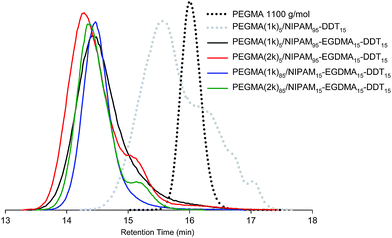 | ||
| Fig. 1 GPC traces (THF, 40 °C, calibration polystyrene standards) of the branched copolymers synthesised in the presence of DDT as the CTA overlaid with the data for the linear polymer PEGMA(1K) and the linear copolymer PEGMA(1K)5/NIPAAm95–DDT15 (synthesised without cross-linker). | ||
In Table 1, we also show data collected using triple detection methods in THF. The measured molecular weights tend to be higher here, but are similar (considering the expected complexity in the architecture) to those measured by single detection. The Mark–Houwink α-values were below 0.32 for all branched copolymers which indicates that all solution morphologies are compact as would be expected from branched architectures as has been found elsewhere.25,28 Further, data collected by single detection methods in DMF were found to be consistent to those measured in THF by single and triple detection methods. These data are also included in Table 1.
Aqueous solution behaviour
The solubility and LCST behaviour of pNIPAAm is known to be dependent on the molecular weight of the pNIPAAm29,30 as well as the end groups.16,31–35 For example, attaching hydrophobic alkyl chains to each end of a pNIPAAm chain results in a decrease in the cloud point and the formation of flower-like micelles at 25 °C.31–33 For PEO-b-pNIPAAm block copolymers, turbidimetry demonstrated that the temperature at which the solution became turbid increased as the hydrophilic content of the copolymer increased.7 For high PEO contents, the magnitude of the turbidity increase is low. Hence, for the branched polymers prepared here, it is to be expected that the hydrophobic CTA and the presence of the poly(ethylene glycol) units will affect the LCST behaviour.Here, all the branched polymers were found to be highly soluble in water at least to a concentration of 150 mg mL−1 at room temperature despite the relatively high concentration of hydrophobic end-groups. Normally, pNIPAAm based polymers exhibit an LCST around 32 °C.8 As a result, increasing the temperature of a solution of pNIPAAm (or a copolymer with PEO) results in a dramatic increase in the turbidity around this temperature due to the formation of large aggregates. For the branched polymers prepared here, in general, at a concentration of 1 mg mL−1, on increasing the temperature the solutions remained transparent up to 70 °C (the maximum temperature studied) which implies that no significant microscopic material is forming during the heating process and these branched copolymers remain colloidally stable. However, an aqueous solution of the PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 branched copolymer showed an increase in absorbance above 30 °C, Fig. 2a. The LCST of pNIPAAm is accompanied by an enthalpy change, which can be measured by DSC.11 The DSC for PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 at a concentration of 125 mg mL−1 shows a broad weak endotherm centred around 29 °C, Fig. 2b. This broad endotherm (as opposed to a sharp LCST) may be a result of the polydispersity of these polymers. The other polymers do not exhibit even this weak endotherm, which we ascribe to the lower weight fraction of NIPAAm in these branched polymers.
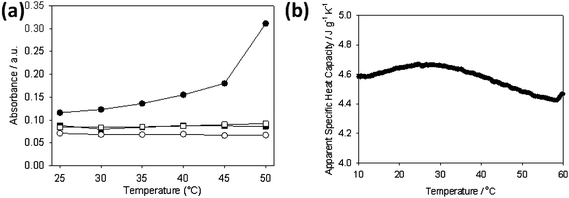 | ||
| Fig. 2 (a) Turbidity data at 350 nm for (●) PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15, (■) PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15, (○) PEGMA(1K)15/NIPAAm85–EGDMA15–DDT15 and (□) PEGMA(2K)15/NIPAAm85–EGDMA15–DDT15 at 10 mg mL−1 in aqueous solution as a function of the temperature between 25 °C and 50 °C. (b) DSC of a 12.5 wt% solution of PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 in water. | ||
Fig. 3 shows an overlay of 1H NMR spectra recorded for the PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 branched copolymer in CDCl3 at 25 °C and in D2O at 25 °C and 60 °C. In CDCl3, the data are very similar to those reported for other PEO–pNIPAAm polymers,36 with additional peaks arising from the DDT. Compared to the data collected in CDCl3, in D2O the well-defined signals from the DDT at 1.2 ppm (–(CH2)9–) and 0.8 ppm (–CH3) were significantly attenuated even at room temperature which implies that these residues are substantially dehydrated and are shielded by the hydrophilic PEGMA (and NIPAAm at 25 °C) residues. Additionally, some attenuation of the backbone protons of the pNIPAAm was observed at 25 °C. Similar attenuation has been shown previously to occur at the LCST of the pNIPAAm for a PEO-b-pNIPAAm copolymer.36 On heating, a slight chemical shift was noticeable between the two spectra recorded in D2O, with some evidence of additional attenuation, although it was possible to observe both the PEGMA and NIPAAm components at 25 °C and 60 °C. As noted above, at 60 °C, the solution was turbid at this temperature, implying that some aggregation events occur under these conditions. Commonly on aggregation of amphiphilic block copolymers, where hydrophobicity drives aggregation, complete attenuation of the hydrophobic block often occurs in the NMR spectrum. The partial attenuation of the NIPAAm observed here could indicate the formation of loose relatively hydrated aggregates.
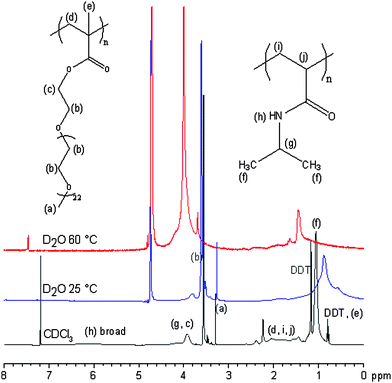 | ||
| Fig. 3 1H NMR spectra recorded for the branched copolymer PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 at 20 mg mL−1 in CDCl3 at 25 °C and D2O at 25 °C and 60 °C. | ||
The temperature-dependent solution behaviour of the branched copolymers at 0.5 mg mL−1 was measured by dynamic light scattering (DLS). The results for the branched copolymers are shown in Fig. 4.
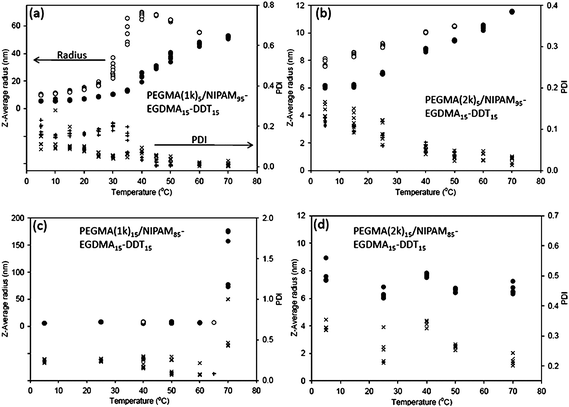 | ||
| Fig. 4 Variation of the intensity z-average radius ● heating; ○ cooling) and the polydispersity index (PDI, × heating; + cooling) with the solution temperature for the branched copolymers (a) PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 (b) PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15 (c) PEGMA(1K)15/NIPAAm85–EGDMA15–DDT15 and (d) PEGMA(2K)15/NIPAAm85–EGDMA15–DDT15 in aqueous solution at a concentration of 0.5 mg mL−1. In all cases, multiple measurements were carried out at each temperature. Some changes with time were observed, hence the differences at any one temperature. | ||
At low temperatures, all branched copolymers were soluble in aqueous solution and displayed hydrodynamic radii between 5 and 10 nm, which is consistent with soluble globular polymer chains of this type. On increasing the temperature, the radius of the particles formed from PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 increased dramatically from 8 nm (5 °C) to 50 nm (70 °C, Fig. 4a). A significant size increase such as this is consistent with a multiple chain self-assembly event. Although this is in agreement with standard block copolymer assembly, where multiple chains aggregate into higher order micellar structures, this observation conflicts with analogous pH-responsive branched copolymer particles which appear to be unimolecular.25 It should be noted that the propensity for chains to assemble and aggregate in an inter-molecular fashion versus their propensity to assemble in an intra-molecular fashion is driven, at least in part, by the balance of the hydrophilic to the hydrophobic component and the ability of the hydrophilic component to stabilise (and thus prevent) inter-molecular association. The precise inter- versus intra-chain assembly will also be composition dependent. Thus, in the current study we probed the effect of both the balance of the hydrophobic (NIPAAm) and hydrophilic (PEGMA) units and the length of the hydrophilic stabilising domains on the intra- versus inter-chain thermally induced assembly. Similarly to the turbidity data (above), the increase in radius observed by DLS was gradual, rather than exhibiting a sharp increase at a well-defined LCST as is often observed for pNIPAAm copolymers. On cooling, a slight increase in size was observed initially, which may be due to continued aggregation with time. Below 40 °C, a decrease in size was observed, before reaching a very similar diameter to the original unheated sample at 5 °C. Thus, the thermal transition appears to be reversible.
In contrast to the short chain PEGMA branched copolymer, PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15 displayed only a modest increase in radius from 6 nm to 11 nm on heating (Fig. 4b). This implies that inter-chain aggregation events are inhibited for these branched copolymers when the steric stabilization potential of the branched copolymer is increased. On cooling, a gradual decrease in radius was observed. Similarly, aqueous solutions of the branched copolymers with higher PEGMA molar incorporation (PEGMA(1K)15/NIPAAm85–EGDMA15–DDT15 and PEGMA(2K)15/NIPAAm85–EGDMA15–DDT15, Fig. 4c and d respectively) showed no detectable change in hydrodynamic diameter on raising the temperature. However, above 70 °C, PEGMA(1K)15/NIPAAm85–EGDMA15–DDT15 showed a dramatic increase in average radius. However, this arises from the generation of bimodal distribution, one with a radius close to the original and one with an apparent large diameter. At this temperature, there is also a loss in data quality and an increase in polydispersity. On cooling just below 70 °C, the original radius was restored. In all cases as noted, the size of the particles after the cooling step differed slightly from the initial size. This difference could be reduced, but not totally removed, by stirring at low temperature for 48 h. Thus it appears that in all but one instance (at the lowest PEGMA molecular weight and molar incorporation) inter-polymer aggregation events are inhibited.
In two cases (PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 and PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15, Fig. 4a and b respectively), the scattering objects exhibit very low polydispersities at high temperature (ca. 0.01). Equally low polydispersities have been reported for self-assembled PEO-b-pNIPAAm copolymers, however, these are synthesised by controlled polymerisation techniques.2 For PEGMA(1k)5/NIPAM95–EGDMA15–DDT15 and PEGMA(2k)5/NIPAM95–EGDMA15–DDT15 branched copolymers, the effect of the polymer concentration on particle diameter was examined by preparing additional solutions at 10 mg mL−1—data at 5 °C to 70 °C are shown in Table 2. The results at 10 mg mL−1 are consistent with those obtained at lower concentration, although slightly larger diameters were measured at higher concentration (although heating rate effects cannot be discounted as a potential explanation2). This concentration dependence on aggregate diameter is, however, not observed for PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15.
| Sample | 5 °C | 70 °C | ||
|---|---|---|---|---|
| z-Average radius/nm | PDI | z-Average radius/nm | PDI | |
| PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 | 8 | 0.126 | 81 | 0.085 |
| PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15 | 6 | 0.104 | 11 | 0.075 |
It is noteworthy that a slight increase of radius—between 6 nm and 12 nm—is observed for the PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15 branched copolymer. This could be due to chain confirmation changes and hydration levels in both the core and the shell or very low-level aggregation on heating. Irrespective of the mode of assembly at elevated temperature, it is clear that inter-polymer association events are dramatically reduced when the PEGMA(2K) chain is used in place of the PEGMA(1K).
To further investigate the aggregation of the polymers, solutions of PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 and PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15 were investigated by cryo-TEM. Solutions (10 mg mL−1) were examined at room temperature and also heated to 70 °C. Representative images are shown in Fig. 5. From these images, it can be seen that at room temperature PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 forms well-defined spherical structures, the dimensions of which are in agreement with the DLS data. On warming, this branched polymer forms ill-defined diffuse structures. On the other hand, PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15 forms small spherical structures at room temperature which is again in good agreement with DLS data. On heating, small particles with consistent sizes were detected (albeit at a low level) and are highlighted in Fig. 5d. These data further confirm that the length of the PEGMA chain is important with regards to the aggregation at high temperature. Presumably the longer PEGMA chains better stabilise the structures formed at low temperature and restrict further aggregation as is also observed for many conventional amphiphilic PEO-based block copolymers.
 | ||
| Fig. 5 Top: cryo–TEM images of PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 at (a) room temperature and (b) heated. Bottom: images of PEGMA(2K)5/NIPAAm95–EGDMA15–DDT15 at (c) room temperature and (d) heated. In all cases, the scale bar represents 200 nm. An expansion is shown in the top right of each image. The image size of the expansion is 127 nm (a and b) or 59 nm (c and d). | ||
Temperature-dependant fluorescence
The distinct polarity dependence of the fluorescence emission of pyrene has been widely used to probe the microenvironment of micellar aggregates,37,38 including for pNIPAAm based systems.36,39 In particular, changes in the relative intensities of the first and third vibrational bands (I1 at 372 nm and I3 at 383 nm) are useful in determining whether the pyrene is located within the hydrophobic micelle core or has been released into the aqueous solution. Fig. 6 shows the relative intensity ratios (I1/I3) calculated from the pyrene emission spectra for an aqueous solution of PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 as a function of the solution temperature. Similar to hydrophobically modified alkyl-terminated pNIPAAm polymers,39 the I1/I3 ratio at low temperature is approximately 1.1 (C18-terminated pNIPAAm shows an I1/I3 ratio of approximately 1.2). Similar observations have been made, and rationalized, for analogous pH-responsive branched copolymers where the higher concentrations of hydrophobic end-groups afforded by the branched architectures provide a substantial hydrophobic domain for pyrene irrespective of the hydrophilicity of the responsive units.25 The ratio rises slightly to 1.2 on heating to 60 °C which presumably reflects the increasing hydrophobicity of the NIPAAm at these elevated temperatures. A similarly small increase in the I1/I3 ratio was also observed for the C18-modified pNIPAAm.39 The reason for this slight increase in I1/I3 with an increase in the temperature is unclear. However, the I1/I3 value for pyrene in pure water (i.e. no polymer) has been reported to increase over this temperature range from 1.8 to 2.0.36 Of course, it is difficult to directly compare this increase in magnitude. However, the implication is that a temperature increase can result in an increase in I1/I3. Conversely, data for a PEO-b-pNIPAAm that undergoes a defined LCST transition demonstrates an I1/I3 ratio of 1.7 at 10 °C, decreasing to approximately 1.4 above the LCST.36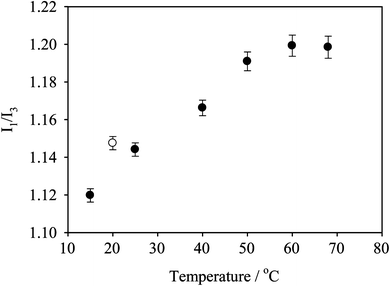 | ||
| Fig. 6 I 1/I3 intensity ratios calculated for the PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 (● heating; ○ cooling) branched copolymer solution at 5 mg mL−1 as a function of the temperature. | ||
Discussion
The data above demonstrate that the branched polymers prepared here do not exhibit conventional LCST behaviour commonly observed for other examples of branched16,17 or star-like40pNIPAAm-based polymers. The only example that demonstrates an increase in turbidity on heating was PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15, however, this increase in turbidity is gradual, rather than being a sharp transition. In addition, NMR and fluorescence measurements show that the PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 exhibits significant hydrophobicity in water even at room temperature. This could suggest that the LCST of the branched copolymer has been lowered below room temperature by the presence of the DDT groups which create a hydrophobic environment when they assemble as has been shown to occur elsewhere for the alkyl-terminated pNIPAAm.31–33 However, even at 5 °C, solutions at 125 mg mL−1 are optically transparent and fully homogeneous. Moreover, DLS studies imply that the structures at low temperature have a radius lower than 10 nm, suggesting that even if inter-polymer aggregation is occurring, the maximum size is inherently limited. The DSC and DLS data imply that there is a broad transition that occurs over a wide temperature range for PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15. This could indicate that the branched copolymer, and particularly the temperature responsive segments of pNIPAAm, undergo a slight change in its hydrophilic nature which is significant enough to trigger the self-assembly of primary particles in large aggregates (the aggregation being due to both the DDT end groups and the NIPAAm). This self-assembly is prevented in the three other cases due to the important amount of the steric stabilizer PEGMA.The lack of a conventional LCST in these polymers gives valuable insight into the structure of these polymers. It has been shown that the LCST of pNIPAAm increases with a decrease in molecular weight29 and that very short oligomers of pNIPAAm do not show any LCST behaviour.30 One explanation for our system is that the absolute length of the pNIPAAm segments within our branched copolymer are very short although we acknowledge that this is difficult to prove unambiguously. From a statistical perspective, this is intuitive since for PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15, the ratio of monomers is 1:19:3:3. Hence, assuming equal reactivity of all of the methacrylates, on average one would expect a ‘chain’ length of approximately three to six NIPAAm units before either a PEGMA, EGDMA or CTA unit is incorporated. Furthermore, the GPC chromatogram of the linear PEGMA(1k)5/NIPAAm95–DDT15 control (Fig. 1) provides clear evidence that the primary chain lengths within the constituent branched copolymers are most likely of very low molecular weight which further reduces the likelihood of successive NIPAAm units being present within these structures and therefore inducing a classical LCST response. A statistical cartoon representation of the PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 branched copolymers, taking only the molar ratio into consideration, is presented in Fig. 7. Several possible structures are drawn, varying only by their molecular weight, with the composition being fixed. We emphasise that the polymerization process is statistical; hence it is highly unlikely that these possible structures are truly representative of the entire mixture and most likely comprise mixtures of each of these. Nonetheless, from this representation it is clear that the choice of the CTA will strongly affect the aqueous solution behaviour of the copolymer. For the branched polymers described here, it can be seen that the chain end is therefore a dominant feature and critical in the assembly of these polymers. To combat the propensity for hydrophobic collapse between CTA end groups and NIPAAm units, high PEGMA contents are required. This differs from the apparent unimicellar behaviour of the pH-responsive equivalents.25
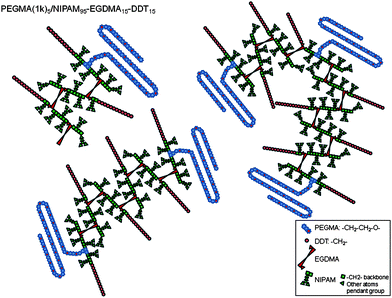 | ||
| Fig. 7 Schematic representation of the branched copolymer PEGMA(1K)5/NIPAAm95–EGDMA15–DDT15 taking only into consideration the molar ratio: several possible structures are proposed, varying by their molecular weight (N.B. this is a cartoon representation which does not reflect the real composition of the sample as free radical polymerisation is likely to lead to heterogeneous samples in terms of the molecular weight and architecture). | ||
Conclusions
We have prepared a series of thermally responsive branched copolymers based on PEGMA and NIPAAm units using a CTA mediated approach. In contrast to pH-responsive branched copolymers25 which behave similarly to analogous individual, discrete nanoparticles such as shell cross-linked micelles, the branched polymers prepared in the study show unique behaviour when compared to PEO-b-pNIPAAm block copolymer micelles. In particular, no conventional LCST behaviour was observed. We attribute this to the low primary chain length and the statistical distribution of the PEGMA, EGDMA and NIPAM units throughout the backbone. Hence, individual NIPAAm sequences tend to be short and, in agreement with recent data, ill-defined LCST transitions. Instead, the aggregation of the polymers appears to be driven by the hydrophobic CTA units which are present at relatively high concentration and to be triggered (in one case) by the NIPAAm units. Increasing the PEGMA chain length restricts this aggregation presumably due to steric bulk. The absence of conventional LCST behavior has, however, provided valuable insight into the structure of these complex architecture polymers. Whilst being seemingly good mimics of pH-responsive micellar systems (where each monomer unit exhibits a pKa), it is clear that the polymer architectures accessed in this study—where there is a highly statistical incorporation of monomer units—may not be suitable for applications which require co-operativity between adjacent monomer units, such as thermo-responsivity. In principle, the preparation of analogous NIPAAm/PEGMA branched copolymers using controlled polymerisation techniques, which allow control of the primary chain length independently from the degree of branching, may provide access to branched vinyl copolymers with conventional LCST transitions.Acknowledgements
We thank the University of Liverpool and the EPSRC (EP/G012741/1) for funding. We thank Professor Steve Armes for access to the DMF-based GPC and Professor Steve Rannard for access to the triple detection GPC. JW thanks the Royal Society for a Royal Society University Research Fellowship.References
- R. X. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627–643 CrossRef CAS.
- J. P. Zhao, G. Z. Zhang and S. Pispas, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4099–4110 CrossRef CAS.
- P. W. Zhu and D. H. Napper, Macromolecules, 1999, 32, 2068–2070 CrossRef CAS.
- J. J. Yan, W. X. Ji, E. Q. Chen, Z. C. Li and D. H. Liang, Macromolecules, 2008, 41, 4908–4913 CrossRef CAS.
- W. Q. Zhang, L. Q. Shi, K. Wu and Y. G. An, Macromolecules, 2005, 38, 5743–5747 CrossRef CAS.
- J. Virtanen, S. Holappa, H. Lemmetyinen and H. Tenhu, Macromolecules, 2002, 35, 4763–4769 CrossRef CAS.
- K. Z. Zhu, R. Pamies, A. L. Kjoniksen and B. Nystrom, Langmuir, 2008, 24, 14227–14233 CrossRef CAS.
- H. W. Chen, J. F. Li, Y. W. Ding, G. Z. Zhang, Q. J. Zhang and C. Wu, Macromolecules, 2005, 38, 4403–4408 CrossRef CAS.
- X. P. Qiu and C. Wu, Macromolecules, 1997, 30, 7921–7926 CrossRef CAS.
- J. Virtanen, C. Baron and H. Tenhu, Macromolecules, 2000, 33, 336–341 CrossRef CAS.
- J. Virtanen and H. Tenhu, Macromolecules, 2000, 33, 5970–5975 CrossRef CAS.
- C. Wu and X. P. Qiu, Phys. Rev. Lett., 1998, 80, 620–622 CrossRef CAS.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS.
- Y. J. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS.
- J. V. M. Weaver and D. J. Adams, Soft Matter, 2010, 6, 2575–2582 RSC.
- S. Carter, B. Hunt and S. Rimmer, Macromolecules, 2005, 38, 4595–4603 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, Macromolecules, 2008, 41, 7368–7373 CrossRef CAS.
- H. Y. Tai, W. X. Wang, T. Vermonden, F. Heath, W. E. Hennink, C. Alexander, K. M. Shakesheff and S. M. Howdle, Biomacromolecules, 2009, 10, 822–828 CrossRef CAS.
- R. M. England and S. Rimmer, Polym. Chem., 2010, 1, 1533 RSC.
- P. A. Costello, I. K. Martin, A. T. Slark, D. C. Sherrington and A. Titterton, Polymer, 2002, 43, 245–254 CrossRef CAS.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef CAS.
- A. T. Slark, D. C. Sherrington, A. Titterton and I. K. Martin, J. Mater. Chem., 2003, 13, 2711–2720 RSC.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, Macromolecules, 2004, 37, 2096–2105 CrossRef CAS.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, J. Mater. Chem., 2003, 13, 2701–2710 RSC.
- J. V. M. Weaver, R. T. Williams, B. J. L. Royles, P. H. Findlay, A. I. Cooper and S. P. Rannard, Soft Matter, 2008, 4, 985–992 RSC.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- S. Gramm, H. Komber and D. Schmaljohann, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 142–148 CrossRef CAS.
- R. T. Woodward and J. V. M. Weaver, Polym. Chem., 2011 10.1039/c0py00277a.
- Y. Xia, N. A. D. Burke and H. D. H. Stover, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- Z. C. Li, Y. H. Kim, H. S. Min, C. K. Han and K. M. Huh, Macromol. Res., 2010, 18, 618–621 Search PubMed.
- R. Nojima, T. Sato, X. P. Qiu and F. M. Winnik, Macromolecules, 2008, 41, 292–294 CrossRef CAS.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 341–348 CrossRef CAS.
- P. Kujawa, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 3048–3055 CrossRef CAS.
- K. Fuchise, R. Kakuchi, S. T. Lin, R. Sakai, S. I. Sato, T. Satoh, W. C. Chen and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6259–6268 CrossRef CAS.
- S. Rimmer, S. Carter, R. Rutkaite, J. W. Haycock and L. Swanson, Soft Matter, 2007, 3, 971–973 RSC.
- R. Motokawa, K. Morishita, S. Koizumi, T. Nakahira and M. Annaka, Macromolecules, 2005, 38, 5748–5760 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- G. B. Ray, I. Chakraborty and S. P. Moulik, J. Colloid Interface Sci., 2006, 294, 248–254 CrossRef CAS.
- H. Ringsdorf, J. Venzmer and F. M. Winnik, Macromolecules, 1991, 24, 1678–1686 CrossRef CAS.
- R. Plummer, D. J. T. Hill and A. K. Whittaker, Macromolecules, 2006, 39, 8379–8388 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |