Connection of polymer chains using diepoxide in CO2/propylene oxide copolymerizations†
Anish
Cyriac
,
Sang Hwan
Lee
and
Bun Yeoul
Lee
*
Department of Molecular Science and Technology, Ajou University, Suwon. 443-749. South Korea. E-mail: bunyeoul@ajou.ac.kr; Fax: + 82-31-219-2394; Tel: + 82-31-219-1844
First published on 21st January 2011
Abstract
When a small amount of vinylcyclohexene dioxide ([diepoxide]/[cat] = 60, 0.14 w% propylene oxide) is additionally added in CO2/propylene oxide copolymerization catalyzed by a highly active catalyst, cobalt(III) complex of a Salen-type ligand tethered by four quaternary ammonium salts (2), some polymer chains are connected consequently resulting in an increased molecular weight of up to Mw = 606000 and broadened molecular weight distribution. The same chain-connecting process is also applicable in the presence of chain transfer agent, tricarballylic acid (C3H5(CO2H)3), 1,2,3,4-butanetetracarboxylic acid (C4H6(CO2H)4), or adipic acid (C4H8(CO2H)2), although a higher amount of diepoxide is needed. Gel permeation chromatography reveals multi-modal distributions along with a tail at a high molecular weight portion due to formation of the connected chains. The shape of the chain transfer agent varies the chain topology, but the rheological properties are not influenced by the topology change. The poly(propylene carbonate)-block-poly(ethylene glycol) copolymer, which is obtained by feeding poly(ethylene glycol)-OH as a chain transfer agent, becomes tough in the tensile properties and shows a high shear sensitivity in the rheological properties when some chains are connected using diepoxide.
Introduction
Carbon dioxide/propylene oxide (PO) alternating copolymer, poly(propylene carbonate) (PPC), has recently drawn great attention from both academic and industrial sectors.1–6 The copolymer is composed of 44% CO2 by weight, burns gently in air without emitting any toxic materials without producing an ash residue, adheres strongly to a cellulosic substrate, and displays a good barrier property for both oxygen and water. We recently reported on a highly efficient copolymerization catalyst (Scheme 1), which allows construction of a pilot plant for continuous process.7,8 The highly active catalyst is a cobalt(III) complex 1, which is prepared from a Salen-type ligand tethered by four quaternary ammonium salts. Catalyst 1 was found while trying to develop a catalyst on the concept of binding two components or two metal centers.9–15 Binding enables the two components or the two metal centers situated in proximity regardless of low catalyst concentration or high polymerization temperature, providing a higher turnover number (TON) and a higher molecular weight (Mn) than the simple binary system of [(Salen)Co(III) or Cr(III) complex]/(onium salt or base)16–18 or the monometallic system.19–21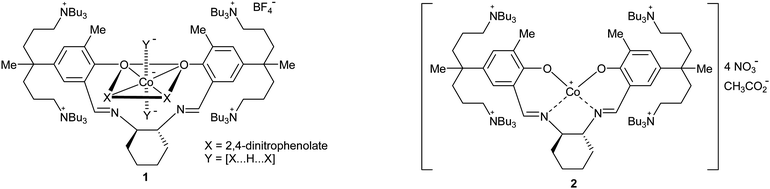 | ||
| Scheme 1 Structure of highly active catalyst for CO2/PO copolymerization. | ||
The extraordinarily high activity of 1 was determined to be due to an unusual binding mode of the cobaltate structure.22–24 The imine-nitrogens on the Salen-type ligand do not coordinate but, instead, the counter anions of the tethered quaternary ammonium coordinate to cobalt. Another advantage of 1 is that the catalyst is efficiently removed after polymerization from a polymer solution through filtration over a short pad of silica gel. The collected catalyst on the silica surface can then be recovered and reused.
The architecture of polymer chains in terms of length, branches, topology, and composition of blocks is a main issue in polymer science. Very recently, precise control of chain length and topology along with architecture of block or graft copolymers with common bulk polymers was realized in the CO2/PO copolymerization through the concept of “immortal polymerization” by feeding various chain transfer agents.25 However, by feeding the chain transfer agents, the molecular weight of the PPC unit is significantly lowered and the molecular weight distribution is too narrow (Mw/Mn, 1.05). In this report, we demonstrate a method not only to increase the molecular weight but also to broaden the molecular weight distribution in CO2/PO copolymerization. Increasing the molecular weight and broadening the molecular weight distribution are key issues in polyolefin industry.26–29 High molecular weight portion is needed to endow mechanical strength, and broad molecular weight distribution enhances the processibility.
Experimental section
General procedure and materials
CO2 gas (99.999% purity) was dried through storage in a column of molecular sieves 3A at a pressure of 30 bar. Propylene oxide (PO) was dried by stirring over CaH2 and then vacuum-transferred to reservoir. The 1H NMR (400 MHz) was recorded on a Varian Mercury Plus 400 apparatus. The gel permeation chromatograms (GPC) were obtained in THF at 35 °C using a Waters Millennium apparatus with polystyrene standards. The Tg data were determined from a second heating at a heating rate of 10 °C min−1 with DSC (Differential Scanning Calorimetry) using Thermal Analysis Q10. The tensile test bars (45 mm × 7.3 mm × 1.2 mm) were prepared by pressing a polymer lump with a screw at 120 °C overnight between two plates that are spaced with a 1.2 mm thick spacer (Fig. 1). The tensile tests were performed according to the ASTM D 638 on UTM (WL2100). The drawing rate was 5 mm/min. Dynamic or oscillatory shear melt rheological data were obtained with a Rheometrics dynamic analyzer model RDAIII using parallel plates in the radian frequency range 0.01–400 s−1. The chain transfer agents and vinylcyclohexene dioxide were purchased from Aldrich. The cobalt(III) complex 2 was prepared as previously described.25 | ||
| Fig. 1 Bar- and coin-shaped samples for the tensile test and the oscillatory shear melt rheological study. | ||
Typical procedure for CO2/PO copolymerization in the presence of diepoxide (entry 7 in Table 1)
A bomb reactor was assembled inside a glove box after being charged with catalyst 2 (3.0 mg, 1.8 μmol), PO (10.5 g, 180 mmol), and vinylcyclohexene dioxide (15 mg, 110 μmol). The reactor was immersed in a 75 °C oil bath. After the solution temperature reached 73 °C (50 min), the CO2 gas was pressurized to 25 bar. The polymerization was performed for 1 h, and a 3–4 bar pressure drop was observed. The reactor was cooled to room temperature through immersion in an ice bath. After CO2 gas was released, the reactor was opened. The polymer solution was filtered over a short pad of silica gel and washed with 10 mL of methylene chloride to give a colorless solution. Any volatiles were removed using a rotary evaporator to give a white residue. In the 1H NMR spectra, cyclic carbonate was observed at less than 5%, which was completely removed by evacuation in a vacuum oven at 150 °C for several hours.| Entry | [Diepoxide]/[2] | TONb | M w c (×10−3) | M w/Mn | T g (°C)d |
|---|---|---|---|---|---|
| a Polymerization conditions: PO (10.5 g, 180 mmol), 2 (3.0 mg, [PO]/[2] = 100000), CO2 (20–25 bar), temperature (70–75 °C), reaction time (60 min). b Turnover number calculated based on the weight of the isolated pure polymers. c Determined on GPC using a polystyrene standard. d Glass transition temperature measured by DSC. e Polymerization time of 90 min. | |||||
| 1 | 0 | 15000 | 257 | 1.37 | 42 |
| 2 | 10 | 15900 | 250 | 1.68 | 42 |
| 3 | 20 | 15300 | 298 | 1.73 | 43 |
| 4 | 30 | 12200 | 325 | 1.96 | 42 |
| 5 | 40 | 13200 | 374 | 2.05 | 43 |
| 6 | 50 | 12400 | 561 | 2.06 | 42 |
| 7 | 60 | 11300 | 606 | 2.11 | 42 |
| 8e | 60 | — | gel | — | — |
| 9 | 70 | — | gel | — | — |
Typical procedure for CO2/PO copolymerization in the presence of diepoxide and chain transfer agent (entry 4 in Table 2)
The polymerization was performed under the same conditions as mentioned above with catalyst 2 (3.0 mg, 1.8 μmol), PO (10.5 g, 180 mmol), tricarballylic acid (5.1 mg, 27 μmol) vinylcyclohexene dioxide (45 mg, 320 μmol).| Entry | Chain transfer agent | [Depoxide]/2 | TON | M w (×10−3) | M w/Mn | T g (°C) |
|---|---|---|---|---|---|---|
| a Polymerization conditions: PO (10.5 g, 180 mmol), 2 (3.0 mg, [PO]/[2] = 100000), tricarballylic acid (5.1 mg, [-CO2H]/[2] = 45), 1,2,3,4-butanetetracarboxylic acid (4.7 mg, [-CO2H]/[2] = 45) or adipic acid (5.9 mg, [-CO2H]/[2] = 45), CO2 (20–25 bar), temperature (70–75 °C), reaction time (60 min). | ||||||
| 1 | C3H5(CO2H)3 | 0 | 13000 | 73 | 1.14 | 40 |
| 2 | C3H5(CO2H)3 | 140 | 14500 | 195 | 2.27 | 40 |
| 3 | C3H5(CO2H)3 | 160 | 14000 | 286 | 2.38 | 41 |
| 4 | C3H5(CO2H)3 | 180 | 13000 | 361 | 2.49 | 42 |
| 5 | C3H5(CO2H)3 | 200 | — | gel | — | — |
| 6 | C4H6(CO2H)4 | 0 | 15500 | 110 | 1.26 | 41 |
| 7 | C4H6(CO2H)4 | 120 | 12000 | 225 | 2.26 | 41 |
| 8 | C4H6(CO2H)4 | 140 | 14400 | 355 | 2.78 | 41 |
| 9 | C4H6(CO2H)4 | 160 | 13400 | 440 | 2.74 | 41 |
| 10 | C4H6(CO2H)4 | 180 | — | gel | — | — |
| 11 | C4H8(CO2H)2 | 0 | 16000 | 76 | 1.12 | 37 |
| 12 | C4H8(CO2H)2 | 200 | 14800 | 165 | 2.06 | 38 |
| 13 | C4H8(CO2H)2 | 220 | 16000 | 181 | 2.20 | 38 |
| 14 | C4H8(CO2H)2 | 240 | 16700 | 363 | 2.83 | 39 |
| 15 | C4H8(CO2H)2 | 260 | — | gel | — | — |
Result and discussion
CO2/PO copolymerization in the presence of diepoxide
When a small amount of diepoxide is additionally added in the CO2/PO copolymerization, the diepoxide is susceptible to incorporation into a polymer chain, resulting in a PPC chain containing some epoxide units. If the growing-chain end reacts with the epoxide unit suspended in another chain, the two growing chains connect by crossing each other (eqn (1)). If this process occurs successively and extensively, the consequence is a gel formation. Actually, when such a small amount of vinylcyclohexene dioxide as 0.01–0.06 mol% (0.024–0.14 w%) with respect to PO is added in the CO2/PO polymerization carried out with catalyst 2, the connected chains are observed in the GPC curves (Fig. 2). Formation of the connected chains results in not only increasing the average molecular weight but also broadening the molecular weight distribution (Table 1). Complex 2, which bears four nitrate anions and an acetate anion instead of 2,4-dinitrophenolate anions in 1, was employed as a catalyst in this study because 2 has some advantages over 1. It can be synthesized in a large scale using inexpensive materials.30 It exhibits an activity (turnover frequency (TOF), ∼15000 h−1) as high as 1 even in the presence of a high amount of protic chain transfer agents.25 As the diepoxide amount increases, the amount of connected chains observed in the gel permeation chromatography (GPC) curve increases (Fig. 2). At a ratio of [PO]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[diepoxide]:[2] = 100000:60:1 (10.5 g PO, 15 mg diepoxide, 3.0 mg catalyst), a high molecular weight of Mw = 606000 is attained with a broad molecular distribution of Mw/Mn = 2.11. The GPC curve is a bimodal distribution centered at ∼200000, which is attributed to unconnected chains, and ∼400000, which is attributed to connected chains. Some tailing is observed at the high molecular weight portions above 1000000, which is attributed to connected regions of many chains (Fig. 2). When the polymerization time is longer (1.5 h instead of 1 h) at the same condition of [diepoxide]/[2] = 60 or when the diepoxide amount increases to [diepoxide]/[2] = 70, a gel is formed. When a gel is formed, the polymerization solution is not flowing and it is unable to remove the catalyst by filtration through a short pad of silica gel. When the remaining unreacted PO is removed by evacuation after gel formation, a highly brittle powdery solid is isolated that is yellowish due to the presence of catalyst residue. If this yellowish powder is kept in an oven at 150 °C for 30 min to completely remove the residual PO, the powder becomes oily due to an unzipping process to form propylene carbonate.31,32 Unless a gel is formed, the polymerization solution is flowing, even though it is very viscous, allowing catalyst removal by filtration over a short pad of silica gel. Once the catalyst is thoroughly removed, the resin is a white and resistant to unzipping process below 200 °C. Glass transition temperature (Tg) and thermal decomposition temperature (Td50) are unaltered at 42 °C and 250 °C, respectively, by the increase of the molecular weight.
:[diepoxide]:[2] = 100000:60:1 (10.5 g PO, 15 mg diepoxide, 3.0 mg catalyst), a high molecular weight of Mw = 606000 is attained with a broad molecular distribution of Mw/Mn = 2.11. The GPC curve is a bimodal distribution centered at ∼200000, which is attributed to unconnected chains, and ∼400000, which is attributed to connected chains. Some tailing is observed at the high molecular weight portions above 1000000, which is attributed to connected regions of many chains (Fig. 2). When the polymerization time is longer (1.5 h instead of 1 h) at the same condition of [diepoxide]/[2] = 60 or when the diepoxide amount increases to [diepoxide]/[2] = 70, a gel is formed. When a gel is formed, the polymerization solution is not flowing and it is unable to remove the catalyst by filtration through a short pad of silica gel. When the remaining unreacted PO is removed by evacuation after gel formation, a highly brittle powdery solid is isolated that is yellowish due to the presence of catalyst residue. If this yellowish powder is kept in an oven at 150 °C for 30 min to completely remove the residual PO, the powder becomes oily due to an unzipping process to form propylene carbonate.31,32 Unless a gel is formed, the polymerization solution is flowing, even though it is very viscous, allowing catalyst removal by filtration over a short pad of silica gel. Once the catalyst is thoroughly removed, the resin is a white and resistant to unzipping process below 200 °C. Glass transition temperature (Tg) and thermal decomposition temperature (Td50) are unaltered at 42 °C and 250 °C, respectively, by the increase of the molecular weight.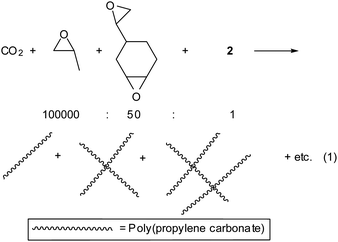 | (1) |
Scheme 2 shows the chain-connecting process by catalyst 2. The five growing polymer chains, which bear an alkoxide or carbonate anion at an end, hang around the cobalt center due to either Columbic interaction between the anion and the ammonium cation or coordination of the anion to the cobalt center.
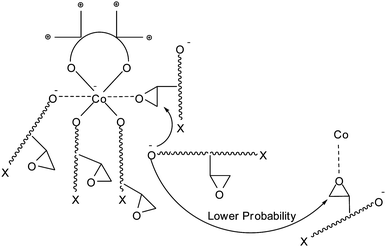 | ||
| Scheme 2 Connecting process of the PPC chains by catalyst 2. | ||
In this situation, connection among the five chains pertained to a cobalt center is more probable than connecting by intermolecular action. This structure of catalysis is beneficial to the connecting process. It may allow for not only some wider operational window before a gel formation but also formation of connected chains even when a minute amount of diepoxide is supplied.
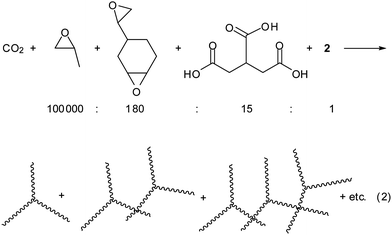 | (2) |
CO2/PO copolymerization in the presence of chain transfer agent and diepoxide
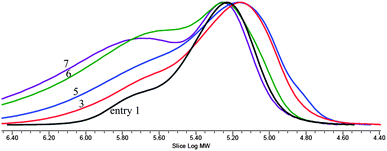
Employing a highly active catalyst 2 and feeding a protic chain transfer agent allows precise control of the chain length and topology in CO2/PO copolymerization.25 Chain transfer reaction between the polymer chains grown from the nitrate or acetate anions in 2 and the fed protic chain transfer agent (alcohol or carboxylic acid) results in liberated polymer chains containing an hydroxyl (–OH) end group and new growing chains. Afterwards, rapid growing-chain swapping occurs through rapid proton exchange reaction between the liberated chains containing neutral –OH end groups and the growing chains bearing carbonate or alcoxide anions at an end. As a consequence, the polymer chains grow uniformly in size from all the added –OH groups and anions in 2 (nitrate and acetate). This kind of chain-transfer polymerization was named “immortal polymerization”.33–35 When tricarballylic acid (C3H5(CO2H)3 bearing three –CO2H groups is added as a chain transfer agent, three armed and star-shaped polymer chains are formed. Especially, when the ratio of [–CO2H]/[2] is as high as 45, the weight fraction grown from the chain transfer agent is 90%, overwhelming those grown linearly from nitrate or acetate anions. The polymer chains grown from the chain transfer agent have –OH groups at all chain ends. In the GPC curve, a narrow main modal is observed centered around 80000 with a minor modal (∼10%) centered around 30000 (entry 1 in Fig. 3). The main modal is attributed to the three-armed and star-shaped chains grown from tricarballylic acid while the minor modal is the chains grown from the nitrate and acetate anions. However, the average molecular weight of the obtained polymer is significantly lowered to 73000 by feeding the chain transfer agent (entry 1 in Table 2).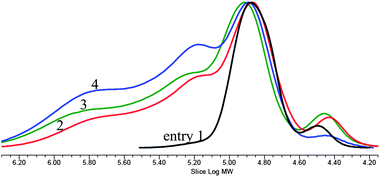
Molecular weight can be increased by additionally feeding diepoxide (eqn (2)). In this case, three times more diepoxide is added to produce the same level of connected chains, because the grown chain length is reduced by one-third from 250000 to 80000 in the presence of the chain transfer agent. Hence, a gel is formed at a [diepoxide]/[2] ratio of 200. In the absence of chain transfer agent, a gel is formed at a [diepoxide]/[2] ratio of 70. At the [diepoxide]/[2] ratio of 180, a significant amount of connected chains is observed in the GPC curve (entry 4 in Fig. 3).
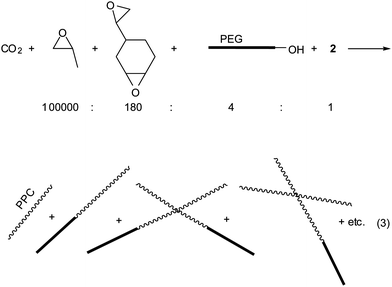 | (3) |
The unconnected three-armed and star-shaped chains are observed as a main modal centered around 80000. At the higher molecular weight region, two more broad modals are observed around 160000 and 600000. The former represents twofold-connected chains. Consequently, the weight-average molecular weight increases from 73000 to 361000, and the molecular weight distribution (Mw/Mn) broadens from 1.14 to 2.49 by feeding diepoxide (entry 4 in Table 2). Through reduction of the fed amount of diepoxide, the amount of connected chains decreases, overall decreasing the average molecular weight and narrowing the molecular weight distribution (entries 2–4 and Fig. 3).
When 1,2,3,4-butanetetracarboxylic acid is fed instead of tricarballylic acid at the same [–CO2H]/[2] ratio of 45, a gel is formed at slightly lower [diepoxide]/[2] ratio of 180 (entry 10 in Table 2). The chain length grown from each –CO2H unit should be same at the same TON, but the overall summed chain length grown from 1,2,3,4-butanetetracarboxylic acid is longer than that grown from tricarballylic acid. Hence, a gel occurs at a lower [diepoxide]/[2] ratio, and more extensive chain connections occur at the same [diepoxide]/[2] ratio (entries 7–9). The GPC curves are similar to those observed following the addition of tricarballylic acid. For the sample in entry 9, main unconnected modal is observed at 130000 with a minor modal at 260000, and another minor modal at 1000000.
When feeding adipic acid at the same [–CO2H]/[2] ratio of 45, a gel is formed at a higher [diepoxide]/[2] ratio of 260 (entry 15 in Table 2). The same-type GPC curve is also observed. For the sample in entry 14, the main unconnected modal is observed at 90000, with minor modals at 200000 and 900000. The attained weight-average molecular weight is in the range of 165000–363000, which are the values attainable in the absence of chain transfer agent without feeding diepoxide. However, the molecular weight distribution and chain topology differ from each other.
CO2/PO copolymerization in the presence of poly(ethylene glycol)-OH and diepoxide
A main demerit of PPC is that mechanically it is easily breakable (brittle) when it is processed to form a film or sheet. This demerit can be overcome by making a block copolymer of PPC-block-poly(ethylene glycol) (PEG).25 The block copolymer is easily prepared by feeding PEG-OH as a chain transfer agent. If the chain length of the fed PEG is sufficiently high (Mn, 35000), the melting signal of the PEG units is observed, even at a low content of PEG units (8.3 w%), at 53 °C along with PPCTg signal in DSC curve. Furthermore, a sheet prepared from the block copolymer is not as brittle as that of PPC itself. It is flexible and stretchable as the conventional PE film.Even though a substantial amount of PEG-OH (250 mg) is fed as a chain transfer reagent, the [–OH]/[2] ratio is low due to the high molecular weight of PEG-OH itself (Mn, 35000). At a target of ∼10 w% PEG with a TON of ∼10000, the [-OH]/[2] ratio is ∼4. In this situation, the obtained polymer is a mixture of chains of PPC itself and those of PPC-block-PEG in an approximate ratio of 1:1. By additionally feeding diepoxide, the molecular weight increases and the molecular weight distribution broadens (entries 2–4 in Table 3, eqn (3)). Under conditions of [diepoxide]/[2] = 180, a molecular weight three times higher (Mw, 225000) and a molecular weight distribution two times broader (Mw/Mn, 2.33) are achieved than those (Mw, 81000; Mw/Mn, 1.20) observed for the sample prepared without feeding diepoxide (entry 4 versus entry 1). In the GPC curves, tailing is observed at the high molecular weight portion.
| Entry | [Depoxide]/2 | TON | w% of PEG | M w (×10−3) | M w/Mn | T g(°C) | T m(°C) |
|---|---|---|---|---|---|---|---|
| a Polymerization conditions: PO (10.5 g, 180 mmol), 2 (3.0 mg, [PO]/[2] = 100000), poly(ethylene glycol (0.25 g, [–OH]/[2] = 4), CO2 (20–25 bar), temperature (70–75 °C), reaction time (60 min). | |||||||
| 1 | 0 | 10300 | 10.2 | 81 | 1.20 | 27 | 53 |
| 2 | 100 | 10800 | 11.2 | 172 | 1.90 | 30 | 53 |
| 3 | 150 | 10800 | 11.2 | 195 | 2.29 | 30 | 53 |
| 4 | 180 | 11000 | 11.0 | 225 | 2.33 | 31 | 54 |
| 5 | 200 | — | — | gel | — | — | — |
Tensile and rheological properties
The tensile strength is significantly increased from 3.0 to 45 N mm−2 due to the increase of the molecular weight (Mw) from 170000 to 606000 through the connection of some PPC chains using diepoxide (entries 1 and 2 in Table 4). However, the brittleness is marginally improved. The high molecular weight PPC containing some connected chains (Mw, 606000) breaks at an elongation of 23% while the low molecular weight PPC (Mw, 170000) breaks at just 7% elongation. In the case of PPC-block-PEG copolymers, the brittleness is significantly improved. The copolymers are stretchable up to above 700% elongation. Through increasing of the molecular weight by the use of diepoxide, a tougher resin is obtained. The tensile strength is increased from 11 to 13 N mm−2 by the increase of molecular weight (Mw) from 81000 to 225000, and the modulus also doubles from 32 to 65 N mm−2 (entries 3 and 4 in Table 3, Fig. 4(A)).![Tensile stress-strain curves (A) and dynamic viscosity curves (B) for the PPC-block-PEG copolymers [a, entry 1 in Table 3 (Mw, 81000; Mw/Mn, 1.20); b, entry 4 in Table 3 (Mw, 225000; Mw/Mn, 2.33)].](/image/article/2011/PY/c0py00365d/c0py00365d-f4.gif) | ||
| Fig. 4 Tensile stress-strain curves (A) and dynamic viscosity curves (B) for the PPC-block-PEG copolymers [a, entry 1 in Table 3 (Mw, 81000; Mw/Mn, 1.20); b, entry 4 in Table 3 (Mw, 225000; Mw/Mn, 2.33)]. | ||
Fig. 4(B) shows the rheological properties of the prepared PPC-block-PEG copolymers (entries 1 and 4 in Table 3). Much higher shear sensitivity is observed for the sample prepared using diepoxide, which may result from either broad molecular weight distribution (Mw/Mn, 2.33) or long chain branches. Polyethylene samples that contain higher amounts of long chain branches display higher shear sensitivity.36 The shear sensitivity is not related to molecular weight itself but is closely correlated with the molecular weight distribution.37 The higher the Mw/Mn value, the higher the shear sensitivity. We encounter many critical processes which require both high viscosity at low shear and low viscosity at high shear. Meeting these requirements involves using a resin with a high degree of shear sensitivity. High melt viscosity at low shear means high melt strength, which is needed in applications such as blow molding and blown films, where excessive drawdown can be a serious problem. Low viscosity at high shear means high flowability, which is needed to minimize friction-generated heat and, consequently, to attain high productivity during processing.
PPC resin containing a substantial amount of connected chains and, hence, showing high molecular weight (Mw, 374000) and broad molecular weight distributions (Mw/Mn, 2.05, entry 5 in Table 1) shows a high shear sensitivity in dynamic viscosity curves (a curve in Fig. 5). However, PPC resins of almost same range of weight-average molecular weights (361000, 363000, and 355000) and somewhat broader molecular-weight distribution (2.49, 2.83, and 2.78 respectively), which are prepared in the presence of both diepoxide and chain transfer agent [tricarballylic acid (entry 4 in Table 2), adipic acid (entry 14 in Table 2), or 1,2,3,4-butanetetracarboxylic acid (entry 8 in Table 2), respectively] show less shear sensitivity than PPC resin prepared in the absence of chain transfer agent (Fig. 5: b, c, and d curves versusa curve).
![Dynamic viscosity curves for the PPCs [a, entry 5 in Table 1 (Mw, 374000; Mw/Mn, 2.05); b, entry 4 in Table 2 (Mw, 361000; Mw/Mn, 2.49); c, entry 14 in Table 2 (Mw, 363000; Mw/Mn, 2.83); d, entry 8 in Table 2 (Mw, 355000; Mw/Mn, 2.78)].](/image/article/2011/PY/c0py00365d/c0py00365d-f5.gif) | ||
| Fig. 5 Dynamic viscosity curves for the PPCs [a, entry 5 in Table 1 (Mw, 374000; Mw/Mn, 2.05); b, entry 4 in Table 2 (Mw, 361000; Mw/Mn, 2.49); c, entry 14 in Table 2 (Mw, 363000; Mw/Mn, 2.83); d, entry 8 in Table 2 (Mw, 355000; Mw/Mn, 2.78)]. | ||
Interestingly, all the three samples prepared in the presence of chain transfer agent show almost the same slope in the fitting of log(complex viscosity) versus log(frequency). In the PPC resin of curve a, some of the high-molecular-weight linear PPC chains are connected, and the connected chains act like the chains containing long chain branches, resulting in a high shear sensitivity (Fig. 5). In the PPC resins of curves b, c, and d, many low-molecular-weight PPC chains, which are linear, or star-shaped, are connected (Fig. 5). In these cases, the connected chains can be better described as the ones containing many short chain branches, not contributing to the shear sensitivity.
Conclusions
A crucial issue in the polyolefin industry is the production of a resin of a broad molecular-weight distribution using either catalyst or process technology.38–42 Typically, a bimodal distribution is advantageous due to a better balance of processability and mechanical properties. A high molecular weight portion is beneficial to the mechanical properties, while a low molecular weight portion is beneficial to the processability. Another important issue in the polyolefin industry is incorporation of long chain branches, which enhance the shear sensitivity in the rheological properties. The resin showing high shear sensitivity is practically advantageous.43 Most of the highly active homogeneous catalysts for CO2/PO copolymerization exhibit a living character in copolymerization, producing PPC with a narrow molecular weight distribution (Mw/Mn, <1.3). Some polymer chains are connected when diepoxide, a vinylcyclohexene dioxide, is additionally added in the CO2/PO copolymerization. Highly active catalyst (TOF > 10000 h−1), cobalt(III) complex of a Salen-type ligand tethered by four quaternary ammonium salts (2), which bears five growing chains around a cobalt center, is beneficial to the connecting process. Feeding a very small amount of diepoxide ([diepoxide]/[2] = 60, 0.14 w% w.r.t. PO) results in a significant amount of connected chains, the consequence of which is increasing the average molecular weight and broadening the molecular weight distribution. The same connecting process can also be applicable in the presence of the chain transfer agents, tricarballylic acid (C3H5(CO2H)3), 1,2,3,4-butanetetracarboxylic acid (C4H6(CO2H)4), and adipic acid (C4H8(CO2H)2), although a higher amount of diepoxide is needed. In the GPC curves, multi-modal distributions are observed along with a tail at the high molecular weight portion due to the connected chains. The shape of the chain transfer agent can vary the chain topology of the resulting PPC, but the rheological properties are not influenced by the topology change. PPC-block-PEG copolymer, which is formed by feeding poly(ethylene glycol)-OH (PEG-OH), becomes tough in the tensile properties and shows high shear sensitivity in the rheological properties, when some chains are connected using diepoxide.Acknowledgements
This work was supported by Inter-Metropolitan Cooperation Development funded by the Presidential Committee on Regional Development and by Construction Technology Innovation Program (CTIP) funded by the MLTM of Korean government.References
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141 RSC.
- G. A. Luinstra, Polym. Rev., 2008, 48, 192 Search PubMed.
- W.-M. Ren, X. Zhang, Y. Liu, J.-F. Li, H. Wang and X.-B. Lu, Macromolecules, 2010, 43, 1396–1402 CrossRef CAS.
- S. I. Vagin, R. Reichardt, S. Klaus and B. Rieger, J. Am. Chem. Soc., 2010, 132, 14367–14369 CrossRef CAS.
- S. S., J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306–7309 CrossRef.
- J. E. Seong, S. J. Na, A. Cyriac, B. W. Kim and B. Y. Lee, Macromolecules, 2010, 43, 903–908 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS.
- B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S.-i. Han, H. Yun, H. Lee and Y.-W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS.
- B. Liu, Y. Gao, X. Zhao, W. Yan and X. Wang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 359–365 CrossRef CAS.
- E. K. Noh, S. J. Na, S. S., S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS.
- D. F. J. Piesik, S. Range and S. Harder, Organometallics, 2008, 27, 6178–6187 CrossRef CAS.
- W.-M. Ren, Z.-W. Liu, Y.-Q. Wen, R. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2009, 131, 11509–11518 CrossRef CAS.
- Y. Xiao, Z. Wang and K. Ding, Chem.–Eur. J., 2005, 11, 3668–3678 CrossRef CAS.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS.
- X.-B. Lu, L. Shi, Y.-M. Wang, R. Zhang, Y.-J. Zhang, X.-J. Peng, Z.-C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664–1674 CrossRef CAS.
- S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 14284–14285 CrossRef CAS.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS.
- B. E. Kim, J. K. Varghese, Y. Han and B. Y. Lee, Bull. Korean Chem. Soc., 2010, 31, 829–834 CrossRef CAS.
- S. J. Na, S. Sujith, A. Cyriac, B. E. Kim, J. Yoo, Y. K. Kang, S. J. Han, C. Lee and B. Y. Lee, Inorg. Chem., 2009, 48, 10455–10465 CrossRef CAS.
- J. Yoo, S. J. Na, H. C. Park, A. Cyriac and B. Y. Lee, Dalton Trans., 2010, 39, 2622–2630 RSC.
- A. Cyriac, S. H. Lee, J. K. Varghese, E. S. Park, J. H. Park and B. Y. Lee, Macromolecules, 2010, 43, 7398–7401 CrossRef CAS.
- P. S. Chum, W. J. Kruper and M. J. Guest, Adv. Mater., 2000, 12, 1759–1767 CrossRef CAS.
- J. Langston, J. Y. Dong and T. C. Chung, Macromolecules, 2005, 38, 5849–5853 CrossRef CAS.
- D. J. Read and J. B. P. Soares, Macromolecules, 2003, 36, 10037–10051 CrossRef CAS.
- O. Sperber and W. Kaminsky, Macromolecules, 2003, 36, 9014–9019 CrossRef CAS.
- J. Min, J. E. Seong, S. J. Na, A. Cyriac and B. Y. Lee, Bull. Korean Chem. Soc., 2009, 30, 745–748 CAS.
- B. Liu, L. Chen, M. Zhang and A. Yu, Macromol. Rapid Commun., 2002, 23, 881–884 CrossRef CAS.
- J. K. Varghese, S. J. Na, J. H. Park, D. Woo, I. Yang and B. Y. Lee, Polym. Degrad. Stab., 2010, 95, 1039–1044 CrossRef CAS.
- S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef CAS.
- K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS.
- H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS.
- R. N. Shroff and H. Mavridis, Macromolecules, 2001, 34, 7362–7367 CrossRef CAS.
- M. Shida and L. V. Cancio, Polym. Eng. Sci., 1971, 11, 124–128 CrossRef CAS.
- F. P. Alt, L. L. Bhm, H. F. Enderle and J. Berthold, Macromol. Symp., 2001, 163, 135–143 CrossRef CAS.
- L. L. Böhm, H. F. Enderle and M. Fleissner, Plast., Rubber Compos. Process. Appl., 1998, 27, 25–29 Search PubMed.
- H. Knuuttila, A. Lehtinen and A. Nummila-Pakarinen, Advances in Polymer Science: Advanced Polyethylene Technologies—Controlled Material Properties, 2004, vol. 169, pp. 13–27 Search PubMed.
- H. T. Liu, C. R. Davey and P. P. Shirodkar, Macromol. Symp., 2003, 195, 309–316 CrossRef CAS.
- K. Yamamoto, Y. Ishihama and K. Sakata, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3722–3728 CrossRef CAS.
- D. Beigzadeh, J. B. P. Soares and T. A. Duever, Macromol. Symp., 2001, 173, 179–194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GPC curves for entries 6–15 in Table 2 and entries 1–4 in Table 4. See DOI: 10.1039/c0py00365d |
| This journal is © The Royal Society of Chemistry 2011 |