Metal catalyzed ring-opening polymerization of benzyl malolactonate: a synthetic access to copolymers of β-benzyl malolactonate and trimethylene carbonate†
Marion
Helou
a,
Guillaume
Moriceau
ab,
Zhi Wei
Huang
b,
Sandrine
Cammas-Marion
b and
Sophie M.
Guillaume
*a
aLaboratoire Catalyse et Organométalliques, CNRS—Université de Rennes 1—Sciences Chimiques de Rennes (UMR 6226), Campus de Beaulieu, 35042, Rennes Cedex, France. E-mail: sophie.guillaume@univ-rennes1.fr; Fax: +33 2 2323 6939; Tel: +33 2 2323 5880
bLaboratoire Chimie Organique et Supramoléculaire, UMR 6226 CNRS, Ecole Nationale Supérieure de Chimie de Rennes (ENSCR), Avenue du Général Leclerc, CS 50837, 35 708, Rennes Cedex 7, France
First published on 12th January 2011
Abstract
The “immortal” coordination–insertion ring-opening polymerization of benzyl malolactonate (MLABe) initiated by the two-component catalyst system based on the zinc amide precursor, (BDI)Zn[N(SiMe3)2] (BDI = β-diiminate ligand), and benzyl alcohol (BnOH) acting as a co-initiator and a chain transfer agent proceeds in bulk at 40 °C. Functional telechelic poly(β-benzyl malolactonate)s, H-PMLABe-OBn, are thus obtained. Sequential copolymerization with trimethylene carbonate (TMC) affords block copolymers, PTMC-b-PMLABe, which are alternatively prepared from the chemical coupling of the PMLABe-COOH and PTMC-OH homopolymers. Simultaneous copolymerization of both the lactone and the carbonate monomers offers the PTMC-co-PMLABe random copolymers. The (co)polymers have been characterized by NMR, FT-IR, SEC and DSC analyses. These represent the first examples of β-benzyl malolactonate/carbonate copolymers. More importantly, these (co)polymers could be synthesized free of metallic residues thereby making them suitable as biomedical and pharmaceutical biomaterials.
Introduction
The impetus of the ongoing research on block copolymers, especially on the biocompatible and biodegradable ones, arises from their potential applications in the biomedical and pharmaceutical fields as temporary therapeutic or diagnostic biomaterials such as drug- or gene-delivery systems or tissue engineering (reparative and regenerative medicines) devices.1 In this regard, copolymers derived from both malic acid (MLA) based polymers, such as poly(alkyl malolactonate)s (PMLARs), or polycarbonates, such as poly(trimethylene carbonate) (PTMC), are of major concern.2,3 Indeed, poly(β-benzyl malolactonate) (PMLABe) is a unique water insoluble functionalized poly(β-alkanoate) which can be easily driven, upon hydrogenation of the benzyl lateral groups, into an hydrophilic polyester. The resulting pendant –COOH groups along the main chain are attractive reactive centers for the immobilization of bioactive molecules (drugs, peptides, proteins).4,5 This versatility of PMLABes makes them highly attractive in the elaboration of nanostructured amphiphilic block copolymers.4b,6 The availability of MLA as various enantiomers or as a racemic mixture, possibly offering each optically active stereoisomers, offers significant interest in modulating the properties of the resulting PMLA. Besides, due to their mechanical softness, PTMC and its copolymers have been investigated as drug delivery vehicles, flexible suture materials, orthopedic screws and pins or implant materials for soft tissue regeneration.2,7,8 The association of both PMLABe and PTMC within a copolymer is thus expected to provide access to the original fully biocompatible and biodegradable nanoobjects with highly valuable thermal and mechanical characteristics and unprecedented physico-chemical behavior. In particular, the introduction of an elastomeric block (PTMC) into a thermoplastic PMLA with tunable crystallinity should allow adjusting the properties of the polymeric material.Regarding the synthesis of poly(β-alkyl malolactonate)s as well as polycarbonates, ring-opening polymerization (ROP) is usually the favored approach over polycondensation.3,9 Indeed, controlled and “living” processes are easily achieved from ROP leading to well defined high molar mass polyesters with controlled molar features (molar mass, molar mass distribution, end-group fidelity) and architectures, and with tailor-made properties.2a,3,4,8 While the synthesis of poly(malic acid) derivatives has been essentially developed via anionic ROP rather than from coordination–insertion ROP,4,5polycarbonates are preferentially prepared via coordination–insertion ROP pathways.2a,3 Among these, we have successfully used the β-diiminate zinc complex (BDI)Zn[N(SiMe3)2] (BDI = CH(CMeNC6H3-2,6-iPr2)2) for the “immortal” ROP (iROP) of TMC.10–12 In association with an alcohol (such as benzyl alcohol, BnOH; for the sake of clarity, the benzyl group “Bn” is distinguished from the benzyl group of the monomer thereafter referred to as “Be”) acting as a co-initiator and a chain transfer agent, this metalloorganic compound, used in truly minute amounts, exhibited unprecedented high activities and productivities in the preparation of well defined PTMCs void of decarboxylation sequences. Furthermore, this same catalytic system has demonstrated its versatility, being successfully applied to the iROP of β-butyrolactone, a four-membered cyclic lactone similar to β-benzyl malolactonate (MLABe) and likewise known for its rather defiant ability to undergo ROP.12,13
In comparison to the copolymers of TMC synthesized with several other cyclic esters,8b,c,14 including with a β-lactone such as the β-butyrolactone comonomer,15copolymers of MLA or its alkyl derivatives (MLARs) with other cyclic esters are quite limited and, to the best of our knowledge, do not include any carbonate moiety.4,5,16–18Block copolymer architectures composed of malic acid or malic acid ester units include poly(malic acid)/poly(lactide),16 poly(malic acid)/poly(β-butyrolactone),16a poly(malic acid)/poly(ε-caprolactone),17 and poly(malic acid)/poly(ethylene glycol).17e,18 Most of these copolymers have been prepared by sequential anionic polymerization of MLAR monomers using potassium alkanoate/18-crown-6, followed by the ROP of the comonomer using a metal-based initiating species like aluminium alkyl17a,c,d or tin (tin(II) bis(2-ethylhexanoate) = Sn(octoate)2 = Sn(oct)2) derivatives.16b,17b,e More scarcely, chain growth of dioxane-dione from poly(lactide) macroinitiators16d or direct copolymerization of both monomers (MLAR and either ε-caprolactone or lactide), using Sn(Oct)216b,17b,e or some organocatalyst(s),16a,d has been carried out. Besides these few examples, as far as we know, no other metal based catalytic system has been used, to date, for the ring-opening (co)polymerization of MLABe. In particular, no single-site metallic system bearing (an) ancillary ligand(s) has allowed the copolymerization of any malic acid based monomer with a cyclic ester (diester, lactone or carbonates).
Herein, we report the homopolymerization of MLABe and its copolymerization with TMC according to an innovative coordination–insertion ROP approach based on the (BDI)Zn[N(SiMe3)2]/BnOH catalytic system. Poly(β-benzyl malolactonate) homopolymers, PMLABe, and poly(β-benzyl malolactonate-b/co-trimethylene carbonate) copolymers, PMLABe-b/co-PTMC, are thus synthesized according to Scheme 1.
![Schematic representation of the synthesis of H-PMLA-OR from the (BDI)Zn[(N(SiMe3)2)]/BnOH or Al(OTf)3/BnOH mediated “immortal” ROP of MLAR.](/image/article/2011/PY/c0py00368a/c0py00368a-s1.gif) | ||
| Scheme 1 Schematic representation of the synthesis of H-PMLA-OR from the (BDI)Zn[(N(SiMe3)2)]/BnOH or Al(OTf)3/BnOH mediated “immortal” ROP of MLAR. | ||
Experimental section
Materials
Manipulations performed under inert atmosphere (argon, <3 ppm of O2) used standard Schlenk, vacuum line and glove-box techniques. Dried and deoxygenated solvents were purified by standard methods and distilled before use. CDCl3 was dried over a mixture of 3 and 4 Å molecular sieves. Racemic-MLABe (MLABe) was synthesized from DL-aspartic acid according to the previously reported synthesis.5gTrimethylene carbonate (TMC, 1,3-dioxane-2-one, Labso Chimie Fine, Blanquefort, France) was first dissolved in THF and stirred over CaH2 for 2 days, before being filtered and dried; TMC was then recrystallized from cold THF. (BDI)Zn(N(SiMe3)2) was synthesized following literature procedures.19Al(OTf)3 (OTf = CF3SO3), benzyl alcohol (C6H5CH2OH = BnOH), dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) were all used as received (Aldrich). Both C6H5C(O)O-PMLABe-C(O)OH5g and H-PTMC-OBn11a,bhomopolymers were prepared as previously reported.Instrumentation and measurements
1H (500 or 200 MHz) and 13C{1H} (125 or 50 MHz) NMR spectra were recorded in CDCl3 on Bruker Avance 400 MHz, AM 500 and DPX 200 spectrometers at 23 °C and were referenced internally by using the residual 1H and 13C solvent resonance relative to tetramethylsilane (δ = 0 ppm).Average molar mass (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n) and molar mass distribution (w/n) values were determined by size-exclusion chromatography (SEC) in THF at 30 °C (flow rate = 1.0 mL min−1) on a Polymer Laboratories PL50 apparatus equipped with a refractive index detector and a PLgel 5 Å Mixed-C column. The polymer samples were dissolved in THF (2 mg mL−1). All elution curves were calibrated with polystyrene standards. nSEC values of PTMCs were calculated using the average correction coefficient previously reported (nSEC = nSEC raw data × 0.73; 0.73 = average of the coefficients determined from low molar mass PTMCs (0.57; n < 5000 g mol−1) and from high molar mass PTMCs (0.88; n > 10
n) and molar mass distribution (w/n) values were determined by size-exclusion chromatography (SEC) in THF at 30 °C (flow rate = 1.0 mL min−1) on a Polymer Laboratories PL50 apparatus equipped with a refractive index detector and a PLgel 5 Å Mixed-C column. The polymer samples were dissolved in THF (2 mg mL−1). All elution curves were calibrated with polystyrene standards. nSEC values of PTMCs were calculated using the average correction coefficient previously reported (nSEC = nSEC raw data × 0.73; 0.73 = average of the coefficients determined from low molar mass PTMCs (0.57; n < 5000 g mol−1) and from high molar mass PTMCs (0.88; n > 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) by using MALDI-ToF-MS and viscosimetry analyses, respectively).8bnSEC values of all other (co)polymers are uncorrected for possible difference in hydrodynamic volume vs.polystyrene standards. The molar mass values of short-chain H-PMLAHe-ORs were determined by 1H NMR analysis from the relative intensities of the signals of PMLAHe chains methylene protons (–CH2OC(O), δ = 3.02 ppm) to those of the chain-end benzyl (OCH2C6H5, δ = 7.38 ppm).
000 g mol−1) by using MALDI-ToF-MS and viscosimetry analyses, respectively).8bnSEC values of all other (co)polymers are uncorrected for possible difference in hydrodynamic volume vs.polystyrene standards. The molar mass values of short-chain H-PMLAHe-ORs were determined by 1H NMR analysis from the relative intensities of the signals of PMLAHe chains methylene protons (–CH2OC(O), δ = 3.02 ppm) to those of the chain-end benzyl (OCH2C6H5, δ = 7.38 ppm).
Monomer conversions were calculated from FTIR spectra (for PMLABe homopolymers) or from 1H NMR spectra of the crude polymer samples by using the integration (Int.) ratio Int.PMLABe/[Int.PMLABe + Int.MLABe] of the methylene group in α-position of the carbonyl (CH2C(O), δ ≈ 2.85 ppm) and Int.PTMC/[Int.PTMC + Int.TMC] using the methylene group in α-position of the carbonate (CH2OC(O), δ ≈ 4.25 ppm).
FTIR spectra of the polymers were acquired on a FTIR Fourier Nicolet 250 apparatus using KBr plates.
Differential scanning calorimetry (DSC) analyses were performed on a Setaram DSC 131 apparatus calibrated with indium at a rate of 10 °C min−1, under a continuous flow of helium (25 mL min−1), using aluminium capsules. Typically, 10 mg of the (co)polymer was weighted in the capsules. The thermograms were recorded according to the following cycles: −40 °C to +180 °C at 10 °C min−1; +180 °C to −40 °C at 10 °C min−1; −40 °C to +180 °C at 10 °C min−1.
Typical MLABe homopolymerization
Typically, (BDI)Zn(N(SiMe3)2) (0.004 g, 6.22 µmol, 1 equiv.) was added to BnOH (3.3 µL, 31.1 µmol, 5 equiv.) placed in toluene (0.1 mL) just prior to the addition of MLABe (0.128 g, 622 µmol, 100 equiv.) (Table 1, entry 4). The mixture was then stirred at 40 °C for the appropriate time (reaction times were not systematically optimized). The reaction was quenched with excess acetic acid (ca. 1 mL of a 16.5 × 10−3 mol L−1 solution in toluene). The resulting mixture was concentrated under vacuum and the conversion determined by 1H NMR analysis of the residue in CDCl3. This crude polymer was then dissolved in CH2Cl2, purified by precipitation in cold methanol, filtered and dried under vacuum at 40 °C overnight (typical isolated yield 90–97%). The final polymer was then analyzed by NMR, FTIR, and SEC (Table 1). 1H NMR data of H-PMLABe-OBn are similar to that already reported (refer to the ESI†).17dH-PMLAHe-OBn was similarly synthesized from the ROP of MLAHe monomer (previously synthesized according to the same method as the one used for MLABe monomer using HeOH instead of BnOH)5f,g,6a using (BDI)Zn(N(SiMe3)2)/BnOH. 1H NMR data of H-PMLAHe-OBn are in good agreement with the expected polymer structure. H-PMLAHe-OBn: 1H NMR (500 MHz; CD3Cl3; δ (ppm)): 7.38 (s, 5H, OCH2Ph), 5.51 (m, 1nH, CHC(O)OHe), 5.18 (s, 2H, OCH2Ph), 4.16 (t, 2nH, CH2(CH2)4CH3), 3.02 (m, 2nH, CHCH2C(O)), 1.65 (t, 2nH, CH2CH2(CH2)3CH3), 1.32 (m, 6H, (CH2)2(CH2)3CH3), 0.91 (t, 3nH, (CH2)5CH3) (Fig. 3). IR: ν = 1745 cm−1 (νC(O)).| Entry |
[MLABe]0:[(BDI)Zn[N(SiMe3)2]]0:[BnOH]0a |
Reaction timeb/h | Conv.c (%) |
ntheo
d/g mol−1 |
nSEC
e/g mol−1 |
w/nf |
TOF g/h−1 |
|---|---|---|---|---|---|---|---|
| a Monomer and alcohol equiv. relative to [(BDI)Zn[Si(Me3)2]]0. b Reaction times were not necessarily optimized. c Monomer conversion determined by 1H NMR. d Theoretical molar mass value calculated from [MLABe]0/[BnOH]0 × monomer conversion × MMLABe + MBnOH, with MMLABe = 206 g mol−1 and MBnOH = 108 g mol−1. e Experimental molar mass value determined by SECvs.polystyrene standards. f Molar mass distribution value determined from SEC chromatogram traces. g Non-optimized turnover frequency expressed in mol(MLABe) × mol[(BDI)Zn[N(SiMe3)2]]/h−1. h Experiment performed with MLAHe (MMLAHe = 200 g mol−1; MnNMR = 2900 g mol−1). All data are representative of at least duplicated experiments. | |||||||
| 1 | 100:1:0 |
6 | 78 | 16200 |
1760 | 1.46 | 13.0 |
| 2 | 100:1:1 |
3 | 54 | 11230 |
1400 | 1.27 | 18.0 |
| 3 | 100:1:1 |
6 | 67 | 13910 |
1550 | 1.20 | 11.2 |
| 4 | 100:1:5 |
6 | 100 | 4230 | 900 | 1.17 | 16.7 |
| 5h | 100:1:5 |
6 | 80 | 3310 | 2850 | 1.13 | 13.3 |
| 6 | 200:1:1 |
7.5 | 52 | 21530 |
1500 | 1.23 | 13.9 |
| 7 | 200:1:1 |
15 | 78 | 32240 |
1500 | 1.33 | 10.4 |
| 8 | 200:1:5 |
15 | 15 | 1340 | 1000 | 1.11 | 2.0 |
| 9 | 200:1:5 |
24 | 30 | 2580 | 2100 | 1.12 | 2.5 |
| 10 | 200:1:10 |
15 | 40 | 1760 | 1150 | 1.24 | 5.3 |
| 11 | 200:1:10 |
24 | 45 | 1970 | 1500 | 1.28 | 3.8 |
| 12 | 200:1:10 |
63 | 70 | 2990 | 1300 | 1.21 | 2.2 |
| 13 | 500:1:1 |
36 | 39 | 40280 |
1000 | 1.33 | 5.4 |
| 14 | 500:1:1 |
72 | 68 | 70150 |
1200 | 1.25 | 4.7 |
| 15 | 500:1:5 |
72 | 5 | — | — | — | 0.3 |
MLABe/TMC block copolymerization
MLABe/TMC random copolymerization
Typically, (BDI)Zn(N(SiMe3)2) (0.005 g, 7.77 µmol, 1 equiv.) was added to BnOH (4.2 µL, 38.9 µmol, 5 equiv.) placed in toluene (0.1 mL) and stirred over 15 min just prior to the addition of the monomers, TMC (0.159 g, 1.55 mmol, 200 equiv.) and MLABe (0.160 g; 777 µmol, 100 equiv.), and of toluene (0.3 mL) (Table 3, entry 5). After stirring over 16 h at 40 °C, the polymerization was then stopped upon addition of acetic acid (ca. 1 mL of a 16.5 × 10−3 mol L−1 solution in toluene) and dried. After determination of the monomers conversion by 1H NMR, the resulting mixture was dissolved in CH2Cl2, precipitated in cold methanol, filtered and finally dried overnight at 40 °C to afford the copolymer (90% yield). The precipitated copolymer was then analyzed by NMR, FTIR, SEC and DSC (Table 3). 1H NMR data of H-PMLABe-coco-PTMC-OBn are similar to those of H-PMLABe-bb-PTMC-OBn. H-PMLABe-coco-PTMC-OBn: 1H NMR (300 MHz; CDCl3; δ (ppm)): 7.22 (m, (5n + 5)H, CO2CH2Ph-PMLABe, OCH2Ph), 5.43 (m, 1nH, CHCO2CH2Ph-PMLABe), 5.01 (m, (2n + 2)H, CO2CH2Ph-PMLABe, OCH2Ph), 4.10 (m, 4mH, CH2O-PTMC), 2.83 (m, 2nH, CHCH2C(O)-PMLABe), 1.87 (m, 2mH, CH2CH2CH2-PTMC). 13C NMR (75 MHz; CDCl3; δ (ppm)): 168.1 (C(O)-PMLABe), 155.0 (–OC(O)O-PTMC), 128.4 (C6H5-PTMC and -PMLABe), 68.5 (CH-PMLABe), 67.5 (CH2C6H5-PMLABe), 64.6 (CH2CH2CH2-PTMC), 34.3 (CH2-PMLABe), 28.1 (CH2-PTMC). IR: ν = 1745 cm−1 (νC(O)). Tg +19.3 °C (Table 3, entry 7).Coupling reaction of C6H5C(O)O-PMLABe-C(O)OH with H-PTMC-OBn
Dicyclohexylcarbodiimide (DCC, 13.3 × 10−3 mol L−1, 27.5 mg in 10 mL of anhydrous CH2Cl2) and 4-(dimethylamino)pyridine (DMAP, 13.2 × 10−3 mol L−1, 16.8 mg in 10 mL of anhydrous CH2Cl2) solutions were prepared beforehand. C6H5C(O)O-PMLABe-C(O)OH5g (nSEC = 9300 g mol−1, w/n = 1.40; 100 mg dissolved in 1 mL of anhydrous CH2Cl2) was added onto an equimolar solution of H-PTMC-OBn (nSEC = 5700 g mol−1, w/n = 1.60; 63 mg in 1 mL of anhydrous CH2Cl2), under inert atmosphere (Table 4, entry 1). After stirring over 15 min, DCC (1.1 equiv., 883 µL of the previously prepared solution) and DMAP (1 equiv., 810 µL of the previously prepared solution) were added and the resulting mixture stirred over 20 h under reflux at 40 °C. After filtration over Celite and drying, the recovered residue was dissolved in CH2Cl2 and precipitated in cold EtOH. After drying under vacuum at 40 °C, the precipitated copolymer (80% yield) was analyzed by NMR, FTIR, SEC and DSC (Table 4). 1H NMR data of C6H5CH2C(O)O-PMLABe-b-PTMC-OBn (refer to the ESI†) are similar to those of H-PMLABe-bb-PTMC-OBn. IR: ν = 1745 cm−1 (νC(O)). nSEC = 9230 g mol−1, w/n = 1.46.
Results and discussion
Homopolymerization of MLABe
In a first approach, the (“immortal”) ROP of MLABe has been carried out in bulk10,20 at 40 °C using the bi-component catalytic system made of [(BDI)Zn(N(SiMe3)2)] (BDI = CH(CMeNC6H3-2,6-iPr2)2) as the catalyst precursor and benzyl alcohol (BnOH) as the co-initiator and chain transfer reagent (Scheme 1). Monomer-to-zinc loadings were ranging from 100 to 500 and an initial alcohol-to-zinc ratio was varied from 0 up to 10. The most significant results are gathered in Table 1. As demonstrated in our previous studies, implementation of this binary catalyst system successfully and straightforwardly generates, upon in situaminolysis, the corresponding active alkoxide species; this thus no longer requires the prior time-consuming isolation of the initiating species and thereby represents a significant advantage.11–13Metal-amide derivatives, such as the homoleptic [Zn(N(SiMe3)2)3] compound, are known to polymerize lactones, yet in a non-controlled process11–13,21 like the behavior observed in the present work for the ROP of MLABe initiated by [(BDI)Zn(N(SiMe3)2)] (Table 1, entry 1). In the absence of any added alcohol, whereas the β-diiminate zinc precursor showed a moderate activity (78 turnovers in 6 h), the polymerization featured a very broad molar mass distribution value (w/n = 1.46) indicative of significant side reactions and/or of a slow initiation process.
In the presence of one equivalent of BnOH (vs. Zn), the [(BDI)Zn(N(SiMe3)2)] complex thus generated a more active alkoxide species that allowed the conversion of 67 equivalents of the 100 of MLABe introduced within 6 h (Table 1, entry 3). Increasing the MLABe content to 200 equivalents then afforded 78% monomer conversion within 15 h (Table 1, entry 7), an activity (156 turnovers in 15 h) thus comparable to that obtained with half less MLABe (Table 1, entry 3; 67 turnovers in 6 h). At much larger monomer loadings (500 equivalents vs. Zn), 72 h were required to convert 68% of MLABe thereby highlighting a decrease of the activity (Table 1, entry 14; 340 turnovers in 72 h).
In the presence of an excess of BnOH (1 < x = up to 10 equiv. vs. Zn) acting as a chain transfer agent, the [(BDI)Zn(N(SiMe3)2)]/BnOH catalytic system allowed the polymerization of MLABe at various monomer-to-zinc and zinc-to-alcohol ratios with high to quantitative monomer conversions being reached within 6–72 h (Table 1, entries 2–15). Monitoring the progress of the polymerization by 1H NMR spectroscopy showed the decrease of the methylene resonance of the malate unit of the monomer at δ 5.30 ppm, and the concomitant appearance of the corresponding broad singlet of the PMLABe at δ 5.53 ppm.
Analysis of the PMLABes by SEC showed traces all exhibiting a unimodal and symmetrical peak. The molar mass distribution values (w/n) ranging from 1.11 to 1.33 were quite narrow, taking into account that the polymerizations were performed in bulk.20,22 In comparison, typical values obtained from anionic ROP (in bulk or in solution) vary from 1.01 to 1.80 depending on the initiating system.5,16–18,23 The molar mass distribution values reported in Table 1 probably reflected an initiation slower than the propagation and/or the occurrence of some side processes (transfer and/or inter- and intramolecular transesterification reactions) all along the propagation step, more likely to occur under the solvent-free operating conditions used in the present work as compared to solution process.22 Theoretical molar mass values (ntheo) have been calculated assuming that all the added alcohol molecules contribute to the “immortal” polymerization. The molar mass values determined by SEC (nSEC), although uncorrected for the difference in hydrodynamic radius vs. the polystyrene standards used for the calibration (as generally found in the literature for the related poly(β-butyrolactone)s),13,19 remained lower than the expected values. A similar behavior was already observed for PMALBes synthesized by anionic ROP of MLABe (ntheo = 5500; 9300; 11300 g mol−1vs. nSEC = 6600; 8300; 5200 g mol−1).17a,d,23a Determination of the molar mass of PMLABes by NMR was not possible because of the overlap of the resonances of the chain-end methylene group (OCH2Ph; δ = 5.16 ppm) with that of the side chain methylene groups (CHC(O)OCH2Ph; δ = 5.04 ppm). However, the use of the hexyl substituted malolactonate (MLAHe) allowed the determination of the molar mass by NMR (nNMR = 2900 g mol−1, refer to Experimental section) which was in good agreement with the calculated value established from the MLAHe-to-BnOH ratio (ntheo = 3310 g mol−1, Table 1, entry 5, Fig. 3).
A second feed experiment supported the “living” character of the ROP of MLABe (Fig. 1). Indeed, a PMLABe (nSEC = 1650 g mol−1, w/n = 1.19) was first prepared (40 °C, 4 h; 90% monomer conversion, ntheo = 1550 g mol−1) by ROP of 25 equiv. of MLABe initiated with [(BDI)Zn(N(SiMe3)2)] (1 equiv.) in the presence of 3 equiv. of BnOH. Resuming the polymerization by addition of 25 equiv. of monomer afforded (40 °C, 4 h; 78% monomer conversion, ntheo = 2900 g mol−1) a PMLABe of greater molar mass (nSEC = 2600 g mol−1) and still narrow molar mass distribution (w/n = 1.22).
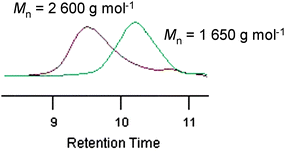 | ||
| Fig. 1
SEC chromatograms of a PMLABe (nSEC = 1650 g mol−1, w/n = 1.19) and a PMLABe (nSEC = 2600 g mol−1, w/n = 1.22). | ||
This [(BDI)Zn(N(SiMe3)2)]0/[BnOH] catalytic system, which proved efficient for the (i)ROP of other cyclic esters such as lactide19 or trimethylene carbonate,11,12 as well as the related four membered ring β-butyrolactone (BBL),12,13 was, however, quite less active for the (i)ROP of MLABe. The activity of the catalytic system was, under the same operating conditions, much larger in the case of BBL than with MLABe: at a ratio [monomer]0/[(BDI)Zn(N(SiMe3)2)]0/[BnOH]0 = 200:1:5, TOFMLABe = 2.5 h−1vs.TOFBBL = 600 h−1.13 Substitution of the β-lactone by the benzyl ester group Be = C(O)OBn rather than by a methyl group thus significantly influenced the polymerization. The presence, on the β-lactone, of a functional group bulkier than Me with a different electronic influence most likely greatly altered the interaction between the catalyst system and the incoming monomer. The anionic ROP of β-substituted β-lactones such as MLABe is classically operated in the presence of a weak base such as tetraalkylammonium benzoate as an initiator.5b,c,g,23 This latter system afforded, upon appropriate purification of MLABe, high molar mass polymers (nSEC up to 174000 g mol−1vs.polystyrene standards in dioxane) with broad molar mass distribution values (1.7 < w/n < 3.3) within 72 h with almost complete monomer conversion (80%).5g In comparison, the present metalloorganic zinc catalytic system showed slightly higher activities (TONzinc up to 18 h−1, Table 1, entry 2, vs. TONanionic = 11 h−1 (ref. 5g)) and significantly narrower molar mass distribution values (w/n ≈ 1.22). This zinc initiator, which operated through a coordination–insertion route,12,13 thus clearly minimized the occurrence of side reactions as supported by NMR analyses (vide infra). This underlines the greater advantages and potential of this new zinc-based catalytic system for the ROP of MLARs.
Some of us recently reported the controlled iROP of TMC from of a two-component catalyst system based on a metal Lewis acid such as a metal triflate Al(OTf)3 (OTf = CF3SO3−) and an alcohol as a co-initiator and a chain transfer agent.24 Similar evaluation of this same catalytic system on the iROP of MLABe and MLAHe in bulk at 40 °C successfully afforded the corresponding PMLARs (Scheme 1). Selected results are reported in Table 2. Both monomers were effectively polymerized under “classical” (1 equiv. of BnOH as a co-initiator) and “immortal” (5 equiv. of BnOH as a co-initiator and chain transfer agent) operating conditions with quite similar to slightly higher activities (100 turnovers in 3–6 h, Table 2) as observed with the above organometallic zinc system (54–100 turnovers in 3–6 h, Table 1, entries 2–5). With this catalytic system, under the operating conditions presently investigated, functionalization of the β-lactone with either a benzyl (Be) or hexyl (He) group does not significantly affect the activities (Table 2). However, this seemed beneficial when compared to the pendant naked β-butyrolactone, BBL, for which significant crotonisation side reactions were observed by NMR analyses under similar bulk conditions12,13 whereas no side reaction was detected in the present case from NMR investigations. The polymerization mechanism in the case of Al(OTf)3/BnOH, based on the activation of the monomer, differs from the coordination–insertion one taking place with the (BDI)Zn[N(SiMe3)2]/BnOH catalytic system, as previously reported.11,12,24
| Entry | Monomer |
[MLAR]0:[M]0:[BnOH]0a |
Reaction timeb/h | Conv.c (%) |
ntheo
d/g mol−1 |
nNMR
e/g mol−1 |
nSEC
f/g mol−1 |
w/nf |
|---|---|---|---|---|---|---|---|---|
| a Monomer and alcohol equiv. relative to [Al(OTf)3]0. b Reaction times were not necessarily optimized. c Monomer conversion determined by 1H NMR. d Theoretical molar mass value calculated from [MLAR]0/[BnOH]0 × monomer conversion × MMLAR + MBnOH, with MMLABe = 206 g mol−1, MMLAHe = 200 g mol−1 and MBnOH = 108 g mol−1. e Experimental molar mass value determined by SECvs.polystyrene standards. f Molar mass distribution value determined from SEC chromatogram traces. All data are representative of at least duplicated experiments. | ||||||||
| 1 | MLABe | 100:1:1 |
6 | 100 | 20700 |
— | 1300 | 1.24 |
| 2 | MLABe | 100:1:5 |
6 | 100 | 4230 | — | 1100 | 1.21 |
| 3 | MLAHe | 100:1:5 |
6 | 100 | 4110 | 6560 | 6400 | 1.20 |
| 4 | MLAHe | 100:1:5 |
3 | 100 | 4110 | 5600 | 5400 | 1.20 |
Copolymers of MLABe and TMC
The catalytic system (BDI)Zn[N(SiMe3)2]/BnOH being demonstrated as efficient for the ROP of both MLABe and TMC,11,12 it was subsequently used to prepare copolymers from these two comonomers (Scheme 2). Copolymers of various compositions were then synthesized upon varying the amount of the TMC and MLABe in the feed and the order of addition of the monomers (Table 3).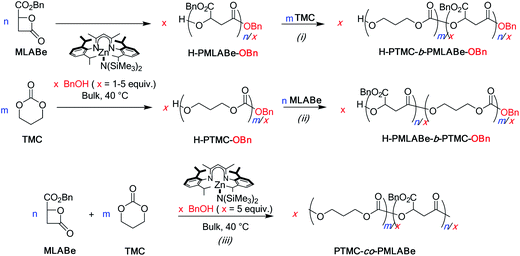 | ||
| Scheme 2 Schematic representation of the syntheses of PMLA-b-/co-PTMC copolymers either from (i and ii) the sequential or (iii) simultaneous copolymerization of MLABe and TMC. | ||
| Entry |
[TMC]0:[MLABe]0:[(BDI)Zn[N(SiMe3)2]]0:[BnOH]0a |
TMC:MLABeb (%) |
Reaction timec | Conv. TMCd (%) | Conv. MLABed (%) | Comp.NMRTMC:MLABee |
ntheo
f/g mol−1 |
nSEC
g/g mol−1 |
w/nh |
|---|---|---|---|---|---|---|---|---|---|
| a Monomer and alcohol equiv. relative to [(BDI)Zn[Si(Me3)2]]0. b Weight fraction of TMC and MLABe units in the feed and relative percentage of TMC and MLABe units in the feed. c Reaction times were not necessarily optimized. d Monomer conversion determined by 1H NMR. e Composition of the precipitated copolymer expressed as weight fraction of TMC and MLABe units in the copolymer; relative percentage of TMC and MLABe units in the copolymer, taking into account the TMC and MLABe conversions determined from 1H NMR analysis. f Theoretical molar mass value calculated from {([TMC]0/[BnOH]0 × Conv.TMC × MTMC) + ([MLABe]0/[BnOH]0 × Conv.MLABe × MMLABe) + MBnOH}, with MTMC = 102 g mol−1, MMLABe = 206 g mol−1 and MBnOH = 108 g mol−1. g Experimental molar mass value determined by SECvs.polystyrene standards. h Molar mass distribution value determined from SEC chromatogram traces. All data are representative of at least duplicated experiments. | |||||||||
| 1 | 200:100:1:5 |
50:50 |
MLABe—6 h + TMC—16 h | 2 | 85 | 2:98 |
3700 | 960 | 1.42 |
| 67:33 |
4:96 |
||||||||
| 2 | 200:100:1:5 |
50:50 |
TMC—3 min + MLABe—16 h | 99 | 88 | 53:47 |
7770 | 7600 | 1.29 |
| 67:33 |
69:31 |
||||||||
| 3 | 100:200:1:5 |
19:81 |
TMC—110 min + MLABe—40 h | 100 | 26 | 49:51 |
4290 | 4850 | 1.37 |
| 33:67 |
67:33 |
||||||||
| 4 | 100:200:1:5 |
19:81 |
TMC—110 min + MLABe—72 h | 100 | 96 | 21:79 |
10060 |
6200 | 1.53 |
| 33:67 |
35:65 |
||||||||
| 5 | 200:100:1:5 |
50:50 |
TMC and MLABe 16 h | 70 | 90 | 44:56 |
6670 | 2450 | 1.62 |
| 67:33 |
61:39 |
||||||||
| 6 | 100:200:1:5 |
19:81 |
TMC and MLABe 48 h | 33 | 93 | 8:92 |
8440 | 2470 | 1.30 |
| 33:67 |
16:84 |
||||||||
| 7 | 100:200:1:5 |
19:81 |
TMC and MLABe 96 h | 78 | 99 | 16:84 |
9860 | 4250 | 1.35 |
| 33:67 |
29:71 |
Sequential copolymerization of MLABe with TMC has been carried out upon introducing both monomers in different order thereby resulting in copolymers with various chain end functions (Scheme 2). Addition of MLABe first followed by TMC (Table 3, entry 1, Scheme 2i) revealed the very slow copolymerization ability of TMC (only 2% conversion of TMC in 16 h) within experimental conditions and especially within the reaction time suitable for complete TMC homopolymerization (100% conversion of 200 equiv. of TMC by [(BDI)Zn[N(SiMe3)2]]0/[BnOH]0 = 1:5 at 60 °C within 5 min).11,12 Under such operating conditions, MLABe/PMLABe thus inhibited the polymerization of TMC. The reverse addition sequence of comonomers, namely the introduction of the benzyl malolactonate once the complete conversion of the carbonate was achieved (i.e., within a few minutes), allowed the synthesis of the desired block copolymer PMLABe-b-PTMC (Table 3, entries 2–4, Scheme 2ii). When TMC was used in excess relative to MLABe (Table 3, entry 2), both monomers were almost fully converted within reaction times close to those expected from their behavior in the corresponding homopolymerizations; TMC was fully converted within 3 min as previously observed11 whereas MLABe was converted slightly more slowly when sequentially copolymerized with TMC (88% within 16 h, Table 3, entry 2) than when present just on its own (100% within 6 h; Table 1, entry 4). When the initial loading of MLABe was increased—to an amount larger than that of TMC (200:100, respectively, Table 3, entries 3 and 4)—the polymerization of the β-substituted β-lactone was slower with only 52 turnovers in 40 h or 192 turnovers in 72 h (Table 3, entries 3 and 4), as expected from the corresponding behavior of MLABe in homopolymerization (60 turnovers in 24 h for 200 equiv. MLABe, Table 1, entry 9, vs. 100 turnovers in 6 h for 100 equiv. MLABe, Table 1, entry 4). SEC analysis of the copolymers showed the increase in molar mass upon going from the PTMC first formed to the subsequent PMLABe-b-PTMC copolymer (Table 3, entry 4; Fig. 2). Such double monomer addition experiments further highlighted the “living” feature of the iROP exemplified above (Fig. 1).
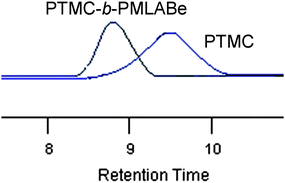 | ||
| Fig. 2
SEC chromatograms of a PTMC (nSEC = 1850 g mol−1, w/n = 1.35) and a PMLABe-b-PTMC (nSEC = 6200 g mol−1, w/n = 1.53) block copolymer obtained by the sequential copolymerization of TMC and MLABe (Table 3, entry 4). | ||
Simultaneous MLABe and TMC addition allowed the synthesis of random copolymers in quite good yields (Table 3, entries 5–7, Scheme 2iii). Whichever the ratio of the comonomers in the feed (TMC/MLABe = 100:200 or 200:100), MLABe always polymerized to a greater extent than TMC, reaching almost quantitative conversions (>90%). TMC, which under similar operating conditions would be fully homopolymerized,11 displayed partial conversion (<78%). The presence of MLABe/PMLABe in the reaction medium thus impeded the polymerization of the carbonate in agreement with the behavior observed in the sequential copolymerization above (Table 3, entry 1). MLABe (200 equiv.) polymerized significantly faster in the presence of TMC (100 equiv.) (Table 3, entry 6; 186 turnovers in 48 h) as compared to its homopolymerization (60 turnovers in 24 h, Table 1, entry 9). TMC was successfully copolymerized with MLABe with high conversion upon increasing the relative ratio of TMC-to-MLABe (Table 3, entry 6 vs. 5) or upon increasing the reaction time (Table 3, entry 6 vs. 7).
The molar composition of the isolated copolymers (Comp.NMR), determined from the integration of the characteristic 1H resonance of PTMC (CH2CH2CH2, δ ≈ 2.05 ppm) and PMLABe (CHC(O)OBe, δ ≈ 5.52 ppm) relative to the corresponding monomer, remained in agreement with the feed ratio (TMC:MLABe). In addition, whatever the composition feed in monomers and the order of their addition, both block and random copolymers exhibited monomodal SEC traces and relatively narrow molar mass distributions (w/n < 1.62).
Alternatively, the block copolymers could be synthesized from the chemical coupling of both pre-synthesized homopolymers, namely carboxylic acid end-capped PMLABe5g and hydroxyl-end-functionalized PTMC11a,b (Scheme 3). A typical esterification between C6H5C(O)O-PMLABe-C(O)OH and H-PTMC-OBn, performed in CH2Cl2 using dicyclohexylcarbodiimide (DCC) as a coupling agent and 4-(dimethylamino)pyridine (DMAP) as a catalyst, as commonly encountered for polyesters,25 afforded after purification and precipitation the block copolymers C6H5C(O)O-PMLABe-b-PTMC-OBn (Table 4). 1H NMR spectrum clearly showed the presence of both blocks (vide infra); yet, it could not demonstrate the effective coupling of these two blocks. This was, however, evidenced from comparative SEC investigations of each homopolymers, of a mixture of both homopolymers and of the coupled homopolymers, i.e. the copolymer itself. Indeed, the chromatogram of a mixture of PMLABe and of PTMC gave two peaks corresponding to each homopolymer (Fig. S1†). Also, the chromatogram of the purified product resulting from their coupling was composed of a unique and distinct peak displaying a quite narrow molar mass distribution (Fig. S1†). This result demonstrated, in combination with NMR data, that the coupling reaction effectively gave access to the expected block copolymer PMLABe-b-PTMC, and not to a mixture of the two homopolymers. The combined NMR, SEC and DSC (vide infra) analyses thus supported the block structure of the copolymer. In addition, such results underlined the availability of both the carboxylic and the hydroxyl-end-functional groups on each homopolymers, for further chemical reaction. Besides they validate such a coupling as an alternative method for the preparation of PMLABe-b-PTMC block copolymers. Noteworthy, this coupling approach to the copolymers also allows the introduction of a selected biologically active molecule at the free chain end of the previously synthesized PMLABe block, this molecule being introduced during the initial anionic ROP of MLABe.5,6
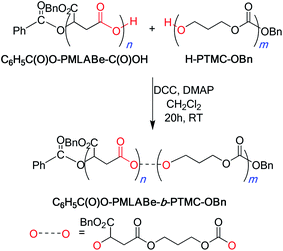 | ||
| Scheme 3 Schematic representation of the synthesis of C6H5C(O)O-PMLABe-b-PTMC-OBn from the direct coupling of C6H5C(O)O-PMLA-C(O)OH and H-PTMC-OBn upon esterification. | ||
| Entry | PMLABe | PTMC | PMLABe-b-PTMC | |||
|---|---|---|---|---|---|---|
|
n
a
|
w/nb |
n
c
|
w/nb |
n
a
|
w/nb |
|
| a Experimental molar mass value determined by SECvs.polystyrene standards. b Molar mass distribution value determined from SEC chromatogram traces. c Experimental molar mass value determined by SECvs.polystyrene standards and corrected by 0.73.8b | ||||||
| 1 | 9300 | 1.40 | 5700 | 1.60 | 9230 | 1.46 |
| 2 | 11100 |
1.34 | 5500 | 1.48 | 6300 | 1.58 |
(Co)polymers characterization
NMR analyses of all the PMLAR homopolymers and their copolymers with PTMC displayed the expected patterns for the main chain signals of both the polyester and polycarbonate moieties. The representative 1H NMR spectra of H-PMLAHe-OBn and PMLABe-b-PTMC (co)polymers are shown in Fig. 3 and 4, respectively. Careful analysis of the chain-end groups of the homopolymers, prepared from the iROP of MLABHe using either bi-component [(BDI)Zn(N(SiMe3)2)] or [Al(OTf)3]/BnOH catalytic systems, invariably revealed the presence of a benzyl terminal group as identified by its diagnostic resonances (Ph-CH2, δ1H = 5.16, δ13C = 70.1 ppm; Ph-CH2, δ1H = 7.38, δ13C = 128 ppm, Fig. 3). This supported the effective role played by the added BnOH as a co-initiator and chain transfer agent in the iROP of MLAR. α-Hydroxy,ω-alkoxyester telechelic linear PMLABe/He was thus unambiguously formed (Scheme 1). Noteworthy, under the operating conditions used and according to the NMR characterization of the precipitated PMLAs (refer to Experimental section), no evidence of any oxygen-alkyl (absence of a carboxy end group at δ 5.6 ppm) nor of any proton abstraction transfer reaction between anionic active sites and polymer chain leading to fumarate groups (absence of any signal at δ 6.8 ppm) could be observed.4,15b,23a This supported the favored oxygen-acyl ring-opening of the monomer. NMR analyses of all copolymers, block, random and coupled ones obtained after precipitation, displayed the signals corresponding to both the PMLA and the PTMC blocks as illustrated with the 1H NMR spectrum of a PMLABe-b-PTMC copolymer (Fig. 4). Notably, in all (co)polymers synthesized, no signals assignable to decarboxylation sequences (absence of the typical signals corresponding to ether units at δ13C = 66.5–67.7 ppm; δ1H = 3.3–3.1 ppm),26 a trend yet often observed in ROP of carbonates,7a were ever observed.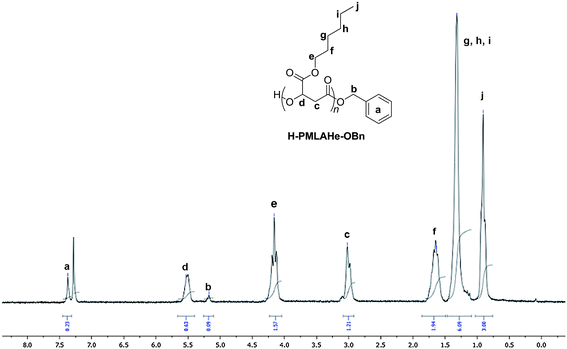 | ||
| Fig. 3 1H NMR (500 MHz, CDCl3, 23 °C) spectrum of a PMLAHe synthesized from the (BDI)Zn(N(SiMe3)2)/BnOH system (Table 1, entry 5). | ||
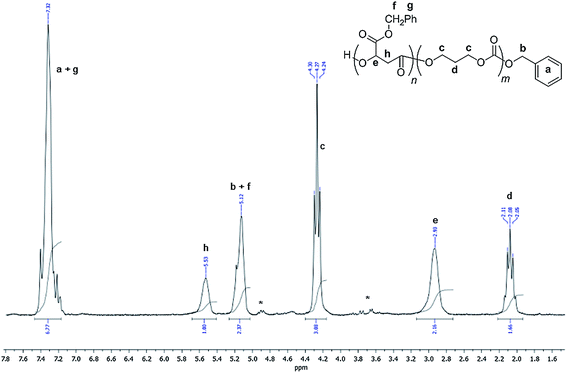 | ||
| Fig. 4 1H NMR (200 MHz, CDCl3, 23 °C) spectrum of a PMLABe-b-PTMC synthesized from the (BDI)Zn(N(SiMe3)2)/BnOH system (Table 3, entry 4) (* refers to residual MLA). | ||
Differential scanning calorimetry (DSC) thermograms of block copolymers exhibit two glass transitions corresponding to each block as illustrated with PMLABe40-b-PTMC20 (Table 3, entry 4; Tg +47.9 °C, −10.5 °C, Fig. 5). No melting temperature was observed in agreement with the racemic feature of the MLABe used and with the amorphous nature of PTMC. These data confirm the block structure of the copolymers prepared from either sequential copolymerization or coupling of the homopolymers. As illustrated with PMLABe40-co-PTMC16, DSC analysis of copolymers obtained through simultaneous copolymerization exhibits a unique glass transition temperature (Tg +19 °C, Table 3, entry 7) intermediate between those of PMLABe (Tg ≈ +29.6 °C, n = 7900 g mol−1)17d and PTMC (Tg ≈ −15 °C, n = 10000 g mol−1)8b in close agreement with the Fox equation, thereby supporting the random structure of the copolymers (Fig. 5).
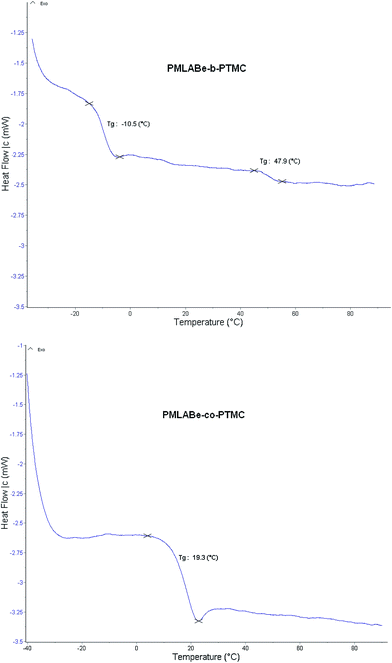 | ||
| Fig. 5 DSC traces of a PMLABe-b-PMTC and PMLABe-co-PMTC (Table 3, entries 4 and 7, respectively; heat flow expressed in mW as a function of temperature expressed in °C; second run). | ||
Conclusion
A new catalytic system based on the β-diiminate zinc complex (BDI)Zn[N(SiMe3)2] associated to BnOH as a co-initiator and a chain transfer agent has been unveiled for the “living” ring-opening polymerization of benzyl and hexyl malolactonate (MLABe and MLAHe). These substituted β-lactones have been successfully homopolymerized under mild reaction conditions (bulk, 40 °C) affording α,ω-hydroxy,alkoxy telechelic poly(β-alkyl malolactonate)s, H-PMLABe/He-OBn. This original coordination–insertion route to poly(β-hydroxyacid) type polyesters allowed the copolymerization of MLABe with TMC. Both block and random copolymers PTMC-b/co-PMLABe were successfully synthesized upon sequential and concomitant addition of the comonomers, respectively. Block copolymers thus obtained exhibited similar characteristics as those prepared upon coupling of the two homopolymers. Thanks to the “immortal” approach and especially given the ease of removal of zinc derivatives,27 such (co)polymers could possibly be synthesized free of metallic residues thereby making them suitable as biomaterials. Thus, such new polycarbonate–poly(alkyl malolactonate) copolymers stand as promising precursors to nanovectors derived from the corresponding amphiphilic polycarbonate–poly(β-malic acid) copolymers.Acknowledgements
This work was financially supported by the Sciences Chimiques de Rennes UMR6226 (grant “Projet inter-équipe 2008” to S.M.G. and S.C.-M.). S.M.G. gratefully thanks Total Petrochemicals Co. (PhD grant to M.H.), Labso Chimie Fine (Blanquefort, France) for kindly supplying TMC, the Region Bretagne ACOMB research program and Rennes Métropole for equipment support. S.C.-M. thanks the Région Bretagne (ARED Polyvect) for PhD grant to Z.W.H.References and notes
- (a) N. Kumar, M. N. V. Ravikumar and A. J. Dombs, Adv. Drug Delivery Rev., 2001, 53, 23–44 CrossRef CAS; (b) S. Li and M. Vert, in The Encyclopedia of Controlled Drug Delivery, ed. E. Mathiowitz, Wiley & Sons, New York, 1999, p. 71 Search PubMed.
- (a) A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS; (b) L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS; (c) T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS; (d) M. Vert, Biomacromolecules, 2005, 6, 538–546 CrossRef CAS.
- (a) A.-C. Albertsson and I. K. Varma, Adv. Polym. Sci., 2002, 157, 1–40 CAS; (b) K. M. Stridsberg, M. Ryner and A.-C. Albertsson, Adv. Polym. Sci., 2002, 157, 41–65 CAS; (c) A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC; (d) C. Jerome and Ph. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef CAS.
- (a) O. Coulembier, P. Degée, J. L. Hedrick and P. Dubois, , Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS; (b) R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- (a) R. Bizzarri, F. Chiellini, R. Solaro, E. Chiellini, S. Cammas-Marion and P. Guerin, Macromolecules, 2002, 4, 1215–1223 CrossRef; (b) K. Boutault, S. Cammas, F. Huet and P. Guerin, Macromolecules, 1995, 28, 3516–3520 CrossRef CAS; (c) L. Moine, S. Cammas, C. Amiel, P. Guerin and B. Sebille, Polymer, 1997, 38, 3121–3127 CrossRef CAS; (d) M. M. Bear, K. Lozac'h, S. Randriamahefa, V. Langlois, R. Bourbouze and P. Guerin, Polymer, 1999, 40, 6251–6528; (e) S. Osanai and K. Nakamura, Biomaterials, 2000, 21, 867–876 CrossRef CAS; (f) S. Cammas-Marion and P. Guérin, Macromol. Symp., 2000, 153, 167–186 CrossRef CAS; (g) S. Cammas, I. Renard, V. Langlois and P. Guérin, Polymer, 1996, 18, 4215–4220 CrossRef.
- (a) S. Cammas, M. M. Béar, A. Harada, Ph. Guérin and K. Kataoka, Macromol. Chem. Phys., 2000, 3, 355–364 CrossRef; (b) M. E. Martinez Barbosa, S. Cammas, M. Appel and G. Ponchel, Biomacromolecules, 2004, 5, 137–143 CrossRef.
- (a) G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS; (b) G. Odian, in Principles of Polymerization, Wiley Interscience, 4th edn, 2004 Search PubMed.
- (a) M. Le Hellaye, N. Fortin, J. Guilloteau, A. Soum, S. Lecommandoux and S. M. Guillaume, Biomacromolecules, 2008, 9, 1924–1933 CrossRef CAS; (b) I. Palard, M. Schappacher, B. Belloncle, A. Soum and S. M. Guillaume, Chem.–Eur. J., 2007, 13, 1511–1521 CrossRef CAS; (c) M. Schappacher, T. Fabre, A. F. Mingotaud and A. Soum, Biomaterials, 2001, 22, 2849–2855 CrossRef.
- K. Pang, R. Kotek and A. Tonelli, Prog. Polym. Sci., 2006, 31, 1009–1037 CrossRef CAS.
- “Immortal” ring-opening polymerization refers to a “living” process in which rapid and reversible chain transfer occurs. S. Penczek and G. Moad, Pure Appl. Chem., 2008, 80, 2163–2193 Search PubMed and ref. 11–13.
- (a) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Chem.–Eur. J., 2008, 14, 8772–8775 CrossRef CAS; (b) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Adv. Synth. Catal., 2009, 351, 1312–1324 CrossRef CAS; (c) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Macromol. Rapid Commun., 2009, 30, 2128–2135 CrossRef CAS; (d) J.-F. Carpentier, S. M. Guillaume, M. Helou, O. Miserque and Y. Sarazin, Eur. Pat. Appl., 08-290187-7, 2008.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- (a) C. Guillaume, J.-F. Carpentier and S. M. Guillaume, Polymer, 2009, 50, 5909–5917 CrossRef CAS; (b) C. Guillaume, N. Ajellal, J.-F. Carpentier and S. M. Guillaume, J. Polym. Sci., Part A: Polym. Chem., 2010 DOI:10.1002/pola.24502.
- (a) Illustrative examples: PTMC–PLA copolymers. D. Pospiech, H. Komber, D. Jehnichen, L. Häussler, K. Eckstein, H. Scheibner, A. Janke, H. R. Kricheldorf and O. Petermann, Biomacromolecules, 2005, 6, 439–446 Search PubMed; (b) Z. Zhang, D. W. Grijpma and J. Feijen, Macromol. Chem. Phys., 2004, 205, 867–875 CrossRef CAS; (c) D. J. Darensbourg, W. Choi, O. Karroonnirum and N. Bhuvanesch, Macromolecules, 2008, 41, 3493–3502 CrossRef CAS ; PTMC–PCL copolymers:; (d) Y. Shen, Z. Shen, Y. Zhang, Q. Huang, L. Shen and H. Yuan, J. Appl. Polym. Sci., 1997, 64, 2131–2139 CrossRef CAS; (e) B. Zhao, C. R. Lu and Q. Shen, J. Appl. Polym. Sci., 2007, 106, 1383–1389 CrossRef CAS; (f) H. Yasuda, M.-S. Aludin, N. Kitamura, M. Tanabe and H. Sirahama, Macromolecules, 1999, 32, 6047–6057 CrossRef CAS ; PTMC–PDMC:; (g) P. Dobrzynski, M. Pastusiak and M. Bero, J. Polym. Sci., 2005, 43, 1913–1922 Search PubMed.
- (a) H. R. Kricheldorf and A. Stricker, Macromol. Chem. Phys., 1999, 200, 1726–1733 CrossRef CAS; (b) Y. Hori, Y. Gonda, Y. Takahashi and T. Hagiwara, Macromolecules, 1996, 29, 804–806 CrossRef CAS.
- (a) O. Coulembier, L. Mespouille, J. L. Hedrick, R. M. Waymouth and P. Dubois, Macromolecules, 2006, 39, 4001–4008 CrossRef CAS; (b) B. He, J. Bei and S. Wang, Polymer, 2003, 44, 989–994 CrossRef CAS; (c) O. Coulembier, P. Degée, P. Guérin and P. Dubois, Langmuir, 2003, 19, 8661–8666 CrossRef CAS; (d) R. J. Pounder and A. P. Dove, Biomacromolecules, 2010, 11, 1930–1939 CrossRef CAS; (e) B. Nottelet, C. Di Tommaso, K. Mondon, R. Gurny and M. Möller, J. Polym. Sci., 2010, 48, 3244–3254 Search PubMed.
- (a) O. Coulembier, P. Degée and P. Dubois, Macromol. Chem. Phys., 2006, 207, 484–491 CrossRef CAS; (b) B. He and M. B. Chan-Park, Macromolecules, 2005, 38, 8227–8234 CrossRef CAS; (c) O. Coulembier, P. Degée, P. Gerbaux, P. Wantier, C. Barbaud, R. Flammang, P. Guérin and P. Dubois, Macromolecules, 2005, 38, 3141–3150 CrossRef CAS; (d) O. Coulembier, P. Degée, S. Cammas-Marion, P. Guérin and P. Dubois, Macromolecules, 2002, 35, 9896–9903 CrossRef CAS; (e) J. Rieger, O. Coulembier, P. Dubois, K. V. Bernaerts, F. E. Du Prez, R. Jérôme and C. Jérôme, Macromolecules, 2005, 38, 10650–10657 CrossRef CAS.
- P. Studer, V. Larras and G. Riess, Eur. Polym. J., 2008, 44, 1714–1721 CrossRef CAS.
- (a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS; (b) L. R. Rieth, D. R. Moore, E. B. Lobkosky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS.
- Taken into account the small amount of solvent used to dissolve the zinc catalyst in the ROP reactions, the procedure is closer to bulk operating conditions rather than to solution ones. It is thus described as bulk process as previously reported.11–13.
- (a) Illustrative examples: Y. Matsuo, K. Mashima and K. Tani, Organometallics, 2001, 20, 3510–3518 Search PubMed; (b) H. Y. Ma and J. Okuda, Macromolecules, 2005, 38, 2665–2673 CrossRef CAS.
- (a) S. Penczek, T. Biela and A. Duda, Macromol. Rapid Commun., 2000, 21, 941–950 CrossRef; (b) S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Slomkowski, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS.
- (a) C. Mabille, M. Masure, P. Hémery and Ph. Guérin, Polym. Bull., 1998, 40, 381–387 CrossRef CAS; (b) A. Hofman, S. Slomkowski and S. Penczek, Makromol. Chem., 1984, 185, 91–101 CrossRef CAS.
- (a) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, ChemCatChem, 2010, 2, 306–313 Search PubMed; (b) J.-F. Carpentier, S. M. Guillaume, M. Helou and O. Miserque, Eur. Pat. Appl., 08-290749-4, 2008.
- (a) Illustrative references: L. Mespouille, O. Coulembier, D. Paneva, P. Degée, I. Raskov and P. Dubois, J. Polym. Sci., 2008, 46, 4997–5013 Search PubMed; (b) H. Li, R. Jerôme and P. Lecomte, Macromolecules, 2008, 41, 650–654 CrossRef CAS; (c) S. Chen, X.-Z. Zhang, S.-X. Cheng, R.-X. Zhuo and Z.-W. Gu, Biomacromolecules, 2008, 9, 2578–2585 CrossRef CAS.
- (a) T. Ariga, T. Takara and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 581–584 CrossRef CAS; (b) S. Agarwal and M. Puchner, Eur. Polym. J., 2002, 38, 2365–2371 CrossRef CAS; (c) S. Agarwal, N. Naumann and X. Xie, Macromolecules, 2002, 35, 7713–7717 CrossRef CAS.
- M. Schappacher, M. Le Hellaye, R. Bareille, M.-C. Durrieu and S. M. Guillaume, Macromol. Biosci., 2010, 10, 60–67 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR data of H-PMLABe-OBn, H-PTMC-b-PMLABe-OBn, and C6H5CH2C(O)O-PMLABe-b-PTMC-OBn, and SEC chromatograms of a mixture of a PMLABe and a PTMC and of the PMLABe-b-PTMC block copolymer obtained by their coupling. See DOI: 10.1039/c0py00368a |
| This journal is © The Royal Society of Chemistry 2011 |