DOI:
10.1039/C0PY00285B
(Paper)
Polym. Chem., 2011,
2, 835-839
Received
3rd September 2010
, Accepted 6th December 2010
First published on 10th January 2011
Abstract
The kinetics of polymerization under confinement is considered. Experimental results show that the polymerization kinetics inside a core–shell electrospun nanofiber differs from that in the bulk (inside a thick film). The observed phenomena can be described in the framework of polychromatic kinetics. In order to explain the chemical reaction mechanism, the spatial features of the process in question, namely the cluster structure forming during the system polymerization, are to be taken into account. The key point of the proposed mechanism is as follows: when the mean size of the clusters becomes comparable with the internal fiber diameter, the rate of reaction will decrease. This decrease is due to two reasons. Firstly, the presence of a boundary obstructs the cluster growth due to steric restrictions; and secondly, the formed clusters separate the reacting area into several isolated regions, in which fluctuations in catalyst concentration are observed without their diffusive dissipation. As a result the polymerization reaction becomes a polychromatic one, where the rate of reaction is decreasing with time.
1. Introduction
Co-electrospinning has been recently proposed for fabricating core–shell fibers made of two different polymer materials.1–6 Such a technique allows one to fabricate new composites on a nanoscale that can provide additional advantages in the physical properties of these materials. Generally, semi-dilute entangled polymer solutions are used in the electrospinning process for the production of nanofibers.7,8 In order to prevent the breakup of the filament into individual droplets the polymer concentration must be several times that of the critical chain overlap concentration, c*.9 However, even for such solution concentrations the volume fraction of the polymer is between 0.1 and 0.2 and the remaining fraction being the solvent. Solidification of the out-flowing jet in the electrospinning occurs in a few milliseconds, where two main stages can be distinguished. First the shell solidifies due to the evaporation in air of the relatively volatile solvent from which the shell solution is composed.10–12 Afterwards, evaporation of the solvent from the core polymer solution continues through the shell by a diffusion process and the core solidifies. Due to the relatively rapid evaporation of the solvent in the core polymer solution, the remained core polymer cannot completely fill the fixed core volume, so the resultant core typically has flaws and a perturbed morphology, and voids exist in the inter-phase between shell and core fibers.13 These flaws will have drastic negative effects on the mechanical properties of core–shell fibers. The above problems can be avoided by using a 100% volume fraction of monomer or oligomer liquid as the core component, with in situpolymerization of the as-spun core–shell fibers. This idea was realized in our previous work.14 The final structure of resin-like polymer matrix of as-spun core–shell nanofibers is formed during polymerization, initiated by UV light in the presence of a photo-initiator. The properties of this structure are substantially being predetermined by the polymerization kinetics, which is the focus of this work.
The key question of this study is: whether the polymerization kinetics under confinement differs from the one in the bulk? If the size-dependent behavior of nanofibers is assumed to be attributed to the confinement of supermolecular structures of nanofiber polymer matrix,15 then it follows that the confinement of the forming structures affects the polymerization kinetics. This assumption is in line with the results of Simon group which has studied the curing behavior of bisphenol M dicyanate ester (BMDC) under the well defined nanoscale constraints imposed by controlled pore glass substrate.16–18 They found that the BMDC physically confined in the pores of controlled pore glass had a higher curing rate than the resin in the bulk state. Similar results were obtained earlier by Amanuel and Malhotra who have studied the effect of confinement on the curing behavior of phenolic resin confined in the nanopores of porous silica substrate.19
2. Materials and methods
Materials
The core–shell fibers were composed of: polycaprolactone (PCL, Mn = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and bisphenol-A-ethoxylate dimethacrylate (BPAEDMA, Mn = 1700) an acrylated aromatic derivative obtained from a condensation reaction of bisphenol A, oligomeric ethyleneoxide, and dimethacrylate. The following solvents were used for solutions preparation: chloroform, dimethylsulfoxide, and 2,2,2-trifluroethanol. As a photoinitiator (PI) 2,2-dimethoxy-2-phenylacetophenone was used. All materials were purchased from Aldrich and used without further purification. PCL was dissolved in a 9:1 (w/w) ratio of chloroform and dimethylsulfoxide to obtain 9 wt% concentrated solution, used as the shell component (shear viscosity 600 cP). As a core component, 2 wt% 2,2-dimethoxy-2-phenylacetophenone dissolved in 98 wt% BPAEDMA was used (shear viscosity 450 cP).
The thermoset polymer fibers were produced using a coaxial electrospinning technique for the fabrication of core–shell fibers (for details, see ref. 2 and 14). PCL was used as the shell, and BPAEDMA with the PI as the core components. The core capillary was a stainless-steel needle with inner and outer diameters of 0.42 and 0.64 mm, respectively. The shell capillary was a plastic needle with inner and outer diameters of 1.2 and 1.75 mm, respectively. The tip of the core capillary protruded between 0.3 and 0.5 mm below that of the shell capillary. The spinning parameters were as follows: shell flow rate was constant at 4 mL h−1, the two different core flow rates were 0.5 and 0.1 mL h−1, electrostatic field was of about 10.5 kV, distance between the spinneret and a collector wheel was 11 cm. The wheel tangential velocity was approximately 20 m s−1. The flow rates of both the core and shell solutions were controlled by two syringe pumps. Typical core diameters of the thin fibers corresponding to the core flow rate of 0.1 mL h−1 were 100/200 nm; and of the thick fibers corresponding to the core flow rate of 0.5 mL h−1 were 250/500 nm (See Fig. 1). All experiments were conducted at room temperature and a relative humidity of 50–60%. BPAEDMA, together with the PI, were encapsulated as the core components, and subsequently post-polymerized by UV radiation. Under the above conditions the oligomer system forms a flexible three dimensional network in a few minutes in both bulk20 and nanofibers.14
000) and bisphenol-A-ethoxylate dimethacrylate (BPAEDMA, Mn = 1700) an acrylated aromatic derivative obtained from a condensation reaction of bisphenol A, oligomeric ethyleneoxide, and dimethacrylate. The following solvents were used for solutions preparation: chloroform, dimethylsulfoxide, and 2,2,2-trifluroethanol. As a photoinitiator (PI) 2,2-dimethoxy-2-phenylacetophenone was used. All materials were purchased from Aldrich and used without further purification. PCL was dissolved in a 9:1 (w/w) ratio of chloroform and dimethylsulfoxide to obtain 9 wt% concentrated solution, used as the shell component (shear viscosity 600 cP). As a core component, 2 wt% 2,2-dimethoxy-2-phenylacetophenone dissolved in 98 wt% BPAEDMA was used (shear viscosity 450 cP).
The thermoset polymer fibers were produced using a coaxial electrospinning technique for the fabrication of core–shell fibers (for details, see ref. 2 and 14). PCL was used as the shell, and BPAEDMA with the PI as the core components. The core capillary was a stainless-steel needle with inner and outer diameters of 0.42 and 0.64 mm, respectively. The shell capillary was a plastic needle with inner and outer diameters of 1.2 and 1.75 mm, respectively. The tip of the core capillary protruded between 0.3 and 0.5 mm below that of the shell capillary. The spinning parameters were as follows: shell flow rate was constant at 4 mL h−1, the two different core flow rates were 0.5 and 0.1 mL h−1, electrostatic field was of about 10.5 kV, distance between the spinneret and a collector wheel was 11 cm. The wheel tangential velocity was approximately 20 m s−1. The flow rates of both the core and shell solutions were controlled by two syringe pumps. Typical core diameters of the thin fibers corresponding to the core flow rate of 0.1 mL h−1 were 100/200 nm; and of the thick fibers corresponding to the core flow rate of 0.5 mL h−1 were 250/500 nm (See Fig. 1). All experiments were conducted at room temperature and a relative humidity of 50–60%. BPAEDMA, together with the PI, were encapsulated as the core components, and subsequently post-polymerized by UV radiation. Under the above conditions the oligomer system forms a flexible three dimensional network in a few minutes in both bulk20 and nanofibers.14
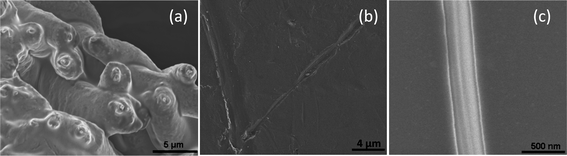 |
| | Fig. 1 (a) Typical image of a core–shell fibers: the core flow rate 0.5 mL h−1, (b) an image of the core fiber after partial UV cross-linking and shell dissolution: exposition time is 3 min that corresponds to beginning of the second stage of the cluster formation when the cluster size is comparable to the inner diameter of the shell and the clusters start to merge, and (c) an image of the core fiber after UV cross-linking (exposition time is 5 min) and shell dissolution. | |
Measurements
Photo-polymerization kinetics was studied by differential scanning photo-calorimeter (METT-LER-DSC 1) equipped with a 200W xenon light source. The wavelength and intensity of the irradiation were 365 nm and 1.12 W cm−2, respectively. All Differential Scanning Calorimetry (DSC) photo-polymerization experiments were repeated five times (with five different samples) for each core flow rate, as well as for the film, under a nitrogen atmosphere (with a 3 min purge interval prior to irradiation) at 25 °C using sample weight ranging between 0.4 and 4 mg. The DSC was calibrated with an indium and zinc melting standard.
3. Results and discussion
Heat flows obtained during photo-polymerization of the BPAEDMA resin encapsulated in PCL electrospun nanofibers are presented in Fig. 2. As the UV radiation starts the heat flow jumps to its maximum and then decreases according to the reaction rate, reaching a plateau as the reaction comes to a stop. After stopping the radiation the heat flow relaxes to the level of the base line. (Note that the base line is slowly increasing with time due to drift.) Thus, the obtained heat flows are formed by the contribution of both the heat released during the polymerization reaction and the heat developed due to the absorption of UV light.20
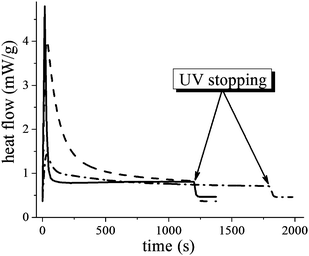 |
| | Fig. 2 Heat flow in DSC experiments for film BPAEDMA/PI (98%/2%) 0.806 mg (solid line); and for nanofibers: thick fibers PCL–BPAEDMA/PI (98%/2%) 3.636 mg, core flow rate is 0.5 mL h−1 (dashed line); thin fibers PCL-BPAEDMA/PI (98%/2%) 0.638 mg, core flow rate is 0.1 mL h−1 (dash-dotted line). | |
Once the heat developed due to UV light absorption is eliminated, one can obtain the portion of the heat flow that is proportional to the polymerization rate (dc(t)/dt), and study the polymerization kinetics corresponding to exponential decay. It turned out that, indeed, the polymerization kinetics under confinement inside a core–shell nanofiber differs from that in the bulk (inside a film) (see Fig. 2). The difference is not only quantitative, but also qualitative; the rate constant of the reaction inside core–shell nanofibers changes during the reaction, unlike the bulk. In the case of only quantitative differences in the polymerization rate, this effect can be attributed to adsorption of UV light in the fiber shell. However, the polychromatic character of the reaction kinetics21,22 observed for the polymerization inside core–shell nanofibers convincingly demonstrates that geometrical confinement of the fiber core substantially affects the chemical reaction (see inset in Fig. 3). The physical mechanism resulting in the polychromatic type of the reaction kinetics can be explained by the model below.
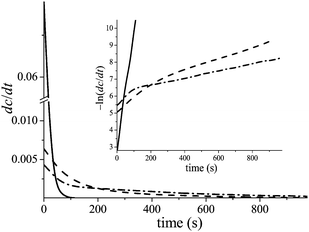 |
| | Fig. 3 Typical kinetic curves of polymerization rate, dc(t)/dt, for film (solid line); and for nanofibers: thick fibers PCL-BPAEDMA/PI (98%/2%) 3.636 mg, core flow rate is 0.5 mL h−1 (dash line); thin fibers PCL-BPAEDMA/PI (98%/2%) 0.638 mg, core flow rate is 0.1 mL h−1 (dash-dot line). The inset demonstrates −ln (dc/dt) vs. time of the above dependences. | |
3.2 Theoretical model of size-dependent polymerization kinetics
In spite of the fact that the oligomer polymerization, initiated by UV light in the presence of a photo-initiator, is a bimolecular reaction, its kinetics corresponds to a reaction of the first order. The point is that each activated radical (the activated end group of an oligomer molecule) can interact with any one of its neighboring molecules, and the probability of this elementary chemical act does not depend on the common concentration of reacting molecules. The reaction constant depends on the photo-initiator concentration, as well as, on the intensity of UV light. Therefore, the polymerization kinetics is to be described by the following equation:| | 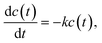 | (1) |
having an exponential solutionwhere k ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) k(pph,IUV) = pphIUVkch is the effective constant of polymerization reaction being proportional to photo-initiator concentration, pph, and intensity of UV light, IUV; and kch is the constant of chemical transformation. Eqn (2) describes well the polymerization kinetics in the bulk (see Fig. 2, solid lines) but is not suitable for a description of the polymerization kinetics inside nanofibers, i.e., under confinement. This fact has a clear explanation: it is impossible to describe a size-dependent phenomenon in the framework of a model containing no parameter for spatial scale. Therefore, a more detailed examination of the polymerization kinetics taking into account spatial scales arising as a result of the polymerization process is required. The simplest way to take into account spatial features of the process in question is to consider the cluster structure forming during the system polymerization. It is reasonable to separate the process into two stages. In the first stage of the cluster formation the dominant process is the cluster initiation. This occurs as a result of the activation of the end groups of the individual oligomer molecules due to a catalyst action under UV light, and their subsequent growth by the addition of individual oligomer molecules to these clusters. In the second stage of the cluster formation, when their mean size is large enough, the clusters start to interact one with each other and begin to merge. Note that in the first stage the concentration of the clusters in the system is increasing, whereas, in the second stage their concentration is decreasing. The key point of such consideration is: when the mean size of the clusters becomes comparable with the internal fiber diameter, the rate of reaction will decrease. The decrease is due to two reasons: the presence of a boundary obstructs the cluster growth due to steric restrictions. Moreover, the formed clusters separate the reacting area into several isolated regions, in which fluctuations of catalyst concentration are observed without their diffusive dissipation. As a result the polymerization reaction becomes polychromatic when the rate of reaction is decreasing with time.21,22 Namely this mechanism can explain the observed behavior in the polymerization process inside the nanofibers.
k(pph,IUV) = pphIUVkch is the effective constant of polymerization reaction being proportional to photo-initiator concentration, pph, and intensity of UV light, IUV; and kch is the constant of chemical transformation. Eqn (2) describes well the polymerization kinetics in the bulk (see Fig. 2, solid lines) but is not suitable for a description of the polymerization kinetics inside nanofibers, i.e., under confinement. This fact has a clear explanation: it is impossible to describe a size-dependent phenomenon in the framework of a model containing no parameter for spatial scale. Therefore, a more detailed examination of the polymerization kinetics taking into account spatial scales arising as a result of the polymerization process is required. The simplest way to take into account spatial features of the process in question is to consider the cluster structure forming during the system polymerization. It is reasonable to separate the process into two stages. In the first stage of the cluster formation the dominant process is the cluster initiation. This occurs as a result of the activation of the end groups of the individual oligomer molecules due to a catalyst action under UV light, and their subsequent growth by the addition of individual oligomer molecules to these clusters. In the second stage of the cluster formation, when their mean size is large enough, the clusters start to interact one with each other and begin to merge. Note that in the first stage the concentration of the clusters in the system is increasing, whereas, in the second stage their concentration is decreasing. The key point of such consideration is: when the mean size of the clusters becomes comparable with the internal fiber diameter, the rate of reaction will decrease. The decrease is due to two reasons: the presence of a boundary obstructs the cluster growth due to steric restrictions. Moreover, the formed clusters separate the reacting area into several isolated regions, in which fluctuations of catalyst concentration are observed without their diffusive dissipation. As a result the polymerization reaction becomes polychromatic when the rate of reaction is decreasing with time.21,22 Namely this mechanism can explain the observed behavior in the polymerization process inside the nanofibers.
Let's consider the first stage of polymerization, when the cluster growth occurs as a result of the addition of individual oligomer molecules to the clusters. This process is described by the following equation:
| | 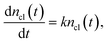 | (3) |
resulting in exponential growth of the clusters.
where
ncl(
t) is the number of individual oligomer molecules which are incorporated into cluster,
ncl(
t = 0) = 1.
The mass of all clusters, formed to the moment of time, t, is
| | 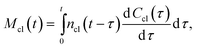 | (5) |
where d
Ccl(
t)/d
t is the rate of new cluster initiation which is described by the following equation:
| | 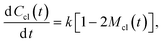 | (6) |
where 1 − 2
Mcl(
t) = [1 −
Mcl(
t)] −
Mcl(
t) =
c(
t) −
Mcl(
t) is the concentration of the individual oligomer molecules with activated end groups which are giving rise to the new clusters, without joining existing clusters.
Substitution of the eqn (4) and (6) into eqn (5) results in the following equation for the function exp (−kt)Mcl(t)
| | 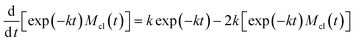 | (7) |
solution of which describes the mass growth of clusters
| | | Mcl(t) = 1 − exp (−kt) | (8) |
where the initial condition
Mcl(
t = 0) = 0 is taken into account. Therefore, the concentration of individual oligomer molecules which are not incorporated into clusters (non-polymerized part of the system) is decreasing according exponential law.
| | | c(t) = 1 − Mcl(t) = exp (−kt) | (9) |
Note that eqn (6), as well as, c(t), eqn (9) are correct only if 1 − 2Mcl(t) > 0, i.e. up to time, t1 ∝ ln(2)/k (at this point in time the concentration of oligomer molecules decreases twice, i.e. it equals to c(t1) ∝ 1/2). Afterwards, the cluster concentration is high and all activated oligomer molecules are immediately being incorporated into existing clusters, without new clusters formation.
Now let's consider the second stage of polymerization, when the cluster joining is dominant. The Smoluchowski kinetic equations are suitable for this goal. The evolution of the individual molecules concentration can be described by the following equation:
| | 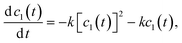 | (10) |
and the evolution of the cluster concentration containing
n individual molecules (
n ≥ 2) can be described by the following equation:
| | 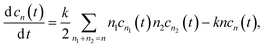 | (11) |
here the rate constant of joining of two cluster of sizes
n1 and
n2 assumes to be equal to
kn1n2, and the conservation law
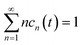
is taken into account.
In the case of cluster–cluster aggregation the concentration of individual oligomer molecules which are not incorporated into clusters (non-polymerized part of the system) is decreasing according to the following law
| | 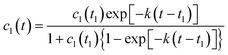 | (12) |
This equation has an exponential asymptote with the same index like in the case of cluster–particle aggregation
| | | c1(t) ∝ exp [−k(t − t1)] | (13) |
Our goal is to estimate only the mean size of the clusters arising during the polymerization. And due to symmetry we do not need to solve eqn (11). Indeed, the mean size of the formed clusters can be calculated as
| | 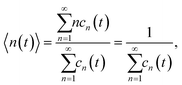 | (14) |
and the equation for the function
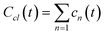
can be obtained by summing the set of
eqn (11) with
eqn (10)| | 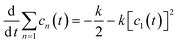 | (15) |
with initial condition
Ccl(
t =
t1) =
Ccl1 which can be obtained from the solution of
eqn (10)| | | Ccl(t) = 2[1 − exp (−kt)] − kt, t < t1, | (16) |
resulting in
Ccl1 = 1 − ln (2).
Thus, the function Ccl(t) equals to
| | 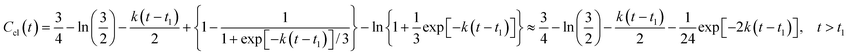 | (17) |
The mean size of the clusters arising during the polymerization equals to
| | 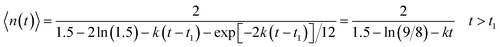 | (18) |
and increases indefinitely at time
t2 which is determined by the following condition
| | | kt2 ≈ 1.5 − ln (9/8) ≈ 1.38 | (19) |
Note that t2 is the upper estimation and in reality the unlimited growth of cluster sizes starts a little earlier.
The above analysis allows one to introduce an additional scale parameter—the cluster size. This scale parameter is to be compared with the diameter of the fiber core. When the mean size of the clusters reaches the diameter of the fiber core, the fiber core will be separated into a large number of slugs consisting of non-polymerized oligomer (see Fig. 1b). This separation occurs earlier than the unlimited increase of the mean size of the clusters begins. The regions separating the slugs of liquid (non-polymerized) oligomer consist of cross-linked (polymerized) molecules. Therefore, mobility in these regions is low, and the diffusive exchange of the catalyst between slugs of non-polymerized oligomer is depressed. It means that after the above separation, the in-homogeneity in catalyst distribution arising due to fluctuation, will not relax. The continued reaction in each slug will occur with a rate corresponding to catalyst concentration in the respective slug. Assuming a uniform distribution of photo-initiator fluctuations with an amplitude Δpph, one can calculate the effective time dependence of concentration of non-polymerized oligomer molecules
| | 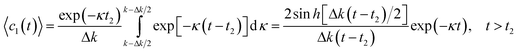 | (20) |
The asymptote of this dependence for
t ≫
t2 corresponds to exponential kinetics with a decreased reaction rate
| | 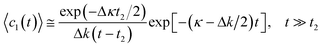 | (21) |
where Δ
k = Δ
pphIUVkch.
Note that in the bulk, even if an infinite cluster is formed, the connectivity between non-reacted regions remains up to a very high level of polymerization, and all fluctuation in catalyst concentration will relax. Therefore, in the bulk no polychromatic kinetics of polymerization reaction will be observed.
The obtained outcomes can be verified by analysis of experimental data. It is clear that the crossover to the polychromatic kinetics should occur before the formation of the infinite cluster. That means that in the experiment the relation ktcr < kt2 should be obtained. Moreover, it is reasonable to assume that the mean size of the cluster achieves the diameter of the fiber core during the second stage of polymerization when the cluster merging is dominant, i.e. tcr > t1. (The cluster mean size at t < t1 is too small.) The experimental data showed in Fig. 2 result in the following values of ktcr satisfying the above inequalities (kt1 < ktcr1; ktcr2 < kt2)
| | | ktcr1 ≈ 0.89, ktcr2 ≈ 1.15, | (22) |
where indices 1 and 2 correspond to the thin and thick fibers, respectively.
In addition, one can predict the difference in the decrease of the slopes of the kinetic curves, for both thin and thick fibers. During the second stage, the number of clusters is decreasing with time, therefore the mean length of the slugs which arise inside the thin fibers is smaller than the mean length of the slugs inside the thick fibers. Thus, the fluctuation amplitude, Δk, of the thin fibers should be higher than that of the thick fibers. And thus the decrease in the slope in the kinetic curves for the thin fibers should be greater than for the thick fibers, and such behavior is observed experimentally (see Fig. 3).
4. Conclusions
Experimental results demonstrated that the polymerization kinetics in core–shell nanofibers differs from the one in bulk not only quantitatively, but also qualitatively. This qualitative difference is important from the point of view of nano-object studies, being additional example of size-dependent behavior. The transformation of the mono-exponential reaction inside a bulk into a polychromatic one inside nanofibers cannot be explained only by the presence of near-surface layers. Even in the thin nanofibers the portion of near-surface layer material does not exceed 2%, whereas the crossover time to the polychromatic kinetics, tcr, corresponds to a degree of polymerization only of about 40%. Therefore, the decrease in reaction rate in the remaining 60% of material cannot be attributed to the effect of the 2% material of near-surface layer. The suggested explanation is based on the analysis of an additional scale parameter arising in the reacting system, namely, the mean size of the clusters forming during the polymerization process. Just the confinement of these clusters results in decrease of the effective reaction rate. This fact lies in line of our concept that confinement of supermolecular structures inside nano-objects causes the modification of their physical and chemical properties.15 In addition, it is important to note that kinetic anomalies can affect the final structure of polymerized system, and that, in turn, can have an impact on the physical properties of the nanofibers.
Acknowledgements
We gratefully acknowledge the financial support of the Russell Berrie Nanotechnology Institute, and the United States—Israel Bi-National Science Foundation (grant 2006061).
References
- D. H. Reneker, A. L. Yarin, E. Zussman and H. Xu, Adv. Appl. Mech., 2007, 41, 43–195 Search PubMed.
- Z. C. Sun, E. Zussman, A. L. Yarin, J. H. Wendorff and A. Greiner, Adv. Mater., 2003, 15, 1929–1936 CrossRef CAS.
- D. Li and Y. Xia, Nano Lett., 2004, 4, 933–938 CrossRef CAS.
- D. Li, J. T. McCann and Y. N. Xia, Small, 2005, 1, 83–86 CrossRef CAS.
- J. H. Yu, S. V. Fridrikh and G. C. Rutledge, Adv. Mater., 2004, 16, 1562–1566 CrossRef CAS.
- Y. Z. Zhang, X. Wang, Y. Feng, J. Li, C. T. Lim and S. Ramakrishna, Biomacromolecules, 2006, 7, 1049–1057 CrossRef CAS.
- J. H. Yu, S. V. Fridrikh and G. C. Rutledge, Polymer, 2006, 47, 4789–4797 CrossRef CAS.
- S. L. Shenoy, W. D. Bates, H. L. Frisch and G. E. Wnek, Polymer, 2005, 46, 3372–3384 CrossRef CAS.
- G. H. McKinley and T. Sridhar, Annu. Rev. Fluid Mech., 2002, 34, 375–415 CrossRef.
- Y. Dror, W. Salalha, R. Avrahami, E. Zussman, A. L. Yarin, R. Dersch, A. Greiner and J. H. Wendorff, Small, 2007, 3, 1064–1073 CrossRef CAS.
- A. Arinstein and E. Zussman, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 056303 CrossRef CAS.
- A. Arinstein, R. Avrahami and E. Zussman, J. Phys. D: Appl. Phys., 2009, 42, 015507 CrossRef.
- E. Zussman, A. L. Yarin, A. V. Bazilevsky, R. Avrahami and M. Feldman, Adv. Mater., 2006, 18, 348–353 CrossRef CAS.
- C. S. Reddy, A. Arinstein, R. Avrahami and E. Zussman, J. Mater. Chem., 2009, 19, 7198–7201 RSC.
- A. Arinstein, M. Burman, O. Gendelman and E. Zussman, Nat. Nanotechnol., 2007, 2, 59–62 CrossRef CAS.
- Y. R. Koh, Q. X. Li and S. L. Simon, Thermochim. Acta, 2009, 492, 45–50 CrossRef CAS.
- Q. X. Li and S. L. Simon, Macromolecules, 2009, 42, 3573–3579 CrossRef CAS.
- Q. X. Li and S. L. Simon, Macromolecules, 2008, 41, 1310–1317 CrossRef CAS.
- S. Amanuel and V. M. Malhotra, J. Appl. Polym. Sci., 2006, 99, 3183–3186 CrossRef CAS.
- E. Amerio, M. Sangermano, G. Colucci, G. Malucelli, M. Messori, R. Taurino and P. Fabbri, Macromol. Mater. Eng., 2008, 293, 700–707 CrossRef CAS.
- V. I. Goldanskii, M. A. Kozhushner and L. I. Trakhtenberg, J. Phys. Chem. B, 1997, 101, 10024–10027 CrossRef CAS.
- Y. A. Berlin, D. O. Drobnitsky and V. V. Kuzmin, J. Chem. Phys., 1994, 100, 3163–3168 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.