DOI:
10.1039/C0PY00362J
(Paper)
Polym. Chem., 2011,
2, 851-861
Multi-end functionalised polymer additives synthesised by living anionic polymerisation—the impact of additive molecular structure upon surface properties
Received
3rd November 2010
, Accepted 9th December 2010
First published on 20th January 2011
Abstract
Numerous applications require specific properties at polymer surfaces that differ from the bulk. Herein we describe the novel synthesis of a series of multi-end functionalised poly(styrene) and poly(isoprene) additives carrying 1 to 3 fluoroalkyl (CF) groups. The additives were prepared by endcapping the living chain ends of polymers prepared via living anionic polymerisation. The resulting polymers have been used as additives to render the surface of polymer films hydrophobic/lipophobic and we have characterised these polymer films using static contact angle measurements with water as the contact fluid. We have found that the additive molecular weight, the number of CF groups, additive concentration and annealing conditions have a significant impact upon the resulting surface properties. Increasing the additive concentration and/or number of CF groups resulted in higher contact angles whereas increasing the molecular weight of additive reduced contact angles and surface hydrophobicity. It has been discovered that these additives undergo rapid adsorption to the surface of a thin film in the time taken to produce the film by spin coating and the result is significantly enhanced surface properties. Annealing polystyrene films above the glass transition temperature revealed some interesting behaviour in so much that it demonstrated that on many occasions it is preferable to anneal films containing very small quantities of additive rather than to simply add greater quantities of additive. In addition to contact angles measurements, Rutherford backscattering (RBS) analysis has been carried out on examples of modified poly(isoprene) films to quantitatively analyse the effect of additive molecular weight and number of fluoroalkyl groups on the near surface elemental composition of the modified thin films and confirming the relationship (described above) between these additive molecular parameters and surface adsorption. Finally, we have described a model which compares the behaviour of the additives in thin films to surfactants in solution.
Introduction
There are frequent circumstances where it is desirable for the properties at the surface of a polymer to be distinctly different from the bulk properties. For example, surface chemistries which promote adhesion, wettability, biocompatibility, chemical resistance and hydrophobicity are attractive for a variety of applications. Low surface energy, highly fluorinated polymer surfaces are particularly appealing in terms of their liquid repellence, chemical inertness and low coefficient of friction and imparting these attributes to a surface can result in the polymers finding use in applications such as anti-fouling finishes, biomedical devices, release coatings and filter media.1 The use of fluorinated polymers to deliver these desirable properties is often impractical given their expense and the difficulty in processing such polymers. Poly(tetrafluoroethylene) (PTFE) for example, has a very high melting point (330 °C) and its melt viscosity is such that it cannot easily be processed viamelt extrusion or injection moulding. However, since in many cases only the surface layer of a product needs to possess these specific properties, a much more efficient approach involves the use of a relatively cheap commodity polymer to provide the desirable, mechanical bulk properties and modification of the surface to introduce the desired surface properties. Examples of current methodologies for fluorinating polymer surfaces include plasma treatments,1–4 wet chemical modification5–7 and the application of polymeric surface coatings,8,9 although all of these approaches tend to suffer from (at least one of) being expensive, restricted to batch processing, safety hazards or the generation of large volumes of solvent waste.
An attractive alternative is the use of fluorinated additives and fluorine containing polymers to create low surface energy properties, although fluorinated polymers themselves, such as PTFE, are highly incompatible with the majority of conventional commodity polymers, making them very difficult to blend. Surface adsorption of additives will be spontaneous if there is an accompanying reduction in the surface energy and it is therefore relatively straightforward to increase the hydrophobicity of polymer surfaces by the use of polymers functionalised with low surface energy fluorocarbon (CF) groups. This is an approach that has been demonstrated previously using fluorine containing polymers of varying structures and with varying degrees of success;10–45 the relative merits and limitations of many of these approaches have been recently reviewed.46 Compatibility of any additive with the matrix (bulk) polymer is crucial. The use of fluorinated block copolymers24 as an additive relies on the surface segregation of the block copolymers to the air–polymer interface and the kinetics of surface segregation comes into play; incompatibility of the fluorinated block (with the bulk) results in unavoidable self-organisation of the block copolymers into large aggregates, which significantly retards surface segregation. Even prolonged annealing above the glass transition temperature is insufficient to allow diffusion of the block copolymers from the bulk to the surface to give uniform coverage. In contrast, it is feasible to rapidly transform surface properties by the incorporation of low molecular weight functionalised additive molecules. However, unless such additives are able to subsequently react with the base polymer, which is rarely the case with many commodity polymers that are chemically inert (poly(ethylene), poly(propylene), poly(ethylene terephthalate), the additives are prone to compromise the bulk physical properties and they may also be removed from the surface viaevaporation, dissolution or wear; hence the long term durability of surface properties will be significantly compromised.
In light of these apparent constraints, an alternative and attractive strategy for modifying surface properties is the use of end functionalised polymer chains which preferentially/spontaneously adsorb to the surface. Hirao42–44 prepared polystyrene polymers by anionic polymerization endcapped with up to 32 C8F17 groups by an iterative methodology that involved many reaction steps. When these endcapped polymers in their pure state—not as additives—were cast into films, it was found that surface properties were (not surprisingly) strongly dependent on the number of CF groups. Furthermore, XPS and static contact angle measurements suggest surface saturation of end-functionalized polymer chains in most cases when the number of CF groups reaches between 8 and 16. Static contact angle measurements of these films when the surface is saturated with CF groups were between 110 and 115° with water and between 53 and 56° with dodecane. More recently we have devised a structure for an end functionalised polymer, with the aim of developing the optimal molecular parameters for efficient surface modification when the functionalised polymers are used as additives in low concentrations. The structure is extremely versatile and consists of two distinct components; a multifunctional head group carrying a pre-determined yet low number of fluoroalkyl groups, to which is attached a pendant polymer chain. This pendant polymer chain is intended to play two roles, firstly to provide long term durability for the modified surface through chain entanglement with the bulk subphase and secondly to compatibilise the highly fluorinated head group with the bulk phase. We have recently shown that spontaneous surface segregation of such additives (carrying up to 4 CF groups) to be a particularly effective approach for the surface functionalisation of polymer thin films when used in low concentration.47–50 Moreover, PTFE like surfaces were achieved with as little as 0.1 wt% of a 5000 g mol−1poly(styrene) additive carrying 3 C8F17groups. We have previously reported the synthesis and characterisation of poly(styrene) and poly(methyl methacrylate) additives synthesised by atom transfer radical polymerisation49 and poly(lactide) synthesised by ring opening polymersation.47 Whilst these successful studies provided insights into the relationship between molecular parameters such as the number of fluoroalkyl groups carried by the additive and surface properties, limited information was obtained pertaining to the impact of the molecular weight of the additive on surface segregation and surface properties—such polymerisation mechanisms do not usually permit a high degree of control over the molecular weight beyond relatively low molecular weights. To overcome this limitation we sought to develop an entirely new methodology for the synthesis of analogous polymer additives by living anionic polymerisation—anionic polymerisation being a truly living polymerisation mechanism capable of exerting control over both the molecular weight and polydispersity index for polymers with molecular weights even in excess of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1. Such control over the polymerisation process has allowed us to carry out an exhaustive study into the influence (and limitation) of molecular weight (and number of CF groups) on surface segregation and surface properties with a view to optimising the molecular parameters of the additive molecules. We herein report the development of a synthetic method for the synthesis of poly(styrene) and poly(isoprene) additives viaanionic polymerisation, with a variety of molecular weights (5000–45000 g mol−1) and carrying 1–3 CFgroups. Surface properties were primarily investigated using static contact angle measurements, although Rutherford backscattering (RBS) analysis was carried out on examples of modified poly(isoprene) films to quantitatively analyse the effect of additive molecular weight and number of fluoroalkyl groups on the near surface elemental composition of the modified thin films—confirming the relationship between these additive molecular parameters and surface adsorption. Finally, comprehensive annealing studies of films containing the poly(styrene) additives revealed some unexpected insight into the relative merits of using increased amounts of additive in a thin film and annealing films containing very small amounts of additive.
000000 g mol−1. Such control over the polymerisation process has allowed us to carry out an exhaustive study into the influence (and limitation) of molecular weight (and number of CF groups) on surface segregation and surface properties with a view to optimising the molecular parameters of the additive molecules. We herein report the development of a synthetic method for the synthesis of poly(styrene) and poly(isoprene) additives viaanionic polymerisation, with a variety of molecular weights (5000–45000 g mol−1) and carrying 1–3 CFgroups. Surface properties were primarily investigated using static contact angle measurements, although Rutherford backscattering (RBS) analysis was carried out on examples of modified poly(isoprene) films to quantitatively analyse the effect of additive molecular weight and number of fluoroalkyl groups on the near surface elemental composition of the modified thin films—confirming the relationship between these additive molecular parameters and surface adsorption. Finally, comprehensive annealing studies of films containing the poly(styrene) additives revealed some unexpected insight into the relative merits of using increased amounts of additive in a thin film and annealing films containing very small amounts of additive.
Experimental
Materials
Benzene (HPLC grade), toluene (HPLC grade), styrene (>99%), isoprene (>99%), hexane (HPLC grade), cyclohexane (HPLC grade) (all Aldrich) were purified, dried and degassed by repeated freeze–pump–thaw cycles over calcium hydride (Aldrich). sec-Butyllithium (1.4 M in cyclohexane), 3,5-di-tert-butyl-4-hydroxytoluene (BHT) (99%), 3,5-dihydroxybenzyl alcohol (99%), anhydrous K2CO3 (99.995%), 18-crown-6 (99%), CBr4 (99%), PPh3 (99%) (all Aldrich), 3-(perfluorooctyl)propanol and 1H,1H,2H,2H-perfluorodecyldimethylchlorosilane (98%, Fluorochem) were used as received. Tetrahydrofuran (THF, Aldrich) was purified by repeated freeze–thaw–pump cycles over sodium wire (Aldrich) and benzophenone (Aldrich) until the solution remained purple. Dichloromethane (DCM) was provided by an in-house solvent purification service.
Measurements
1H NMR analysis was carried out on a Varian VNMRS 700 MHz and Bruker Avance-400 MHz spectrometer using CDCl3 as a solvent. Spectra were referenced to the trace of CHCl3 (7.27 ppm) present in the CDCl3. Molecular weights were determined using triple detection size exclusion chromatography (SEC) on a Viscotek 302 with refractive index, viscosity and light scattering detectors, and 2 × 300 mm PLgel 5 µm mixed C columns. THF was used as the eluent with a flow rate of 1.0 ml per min and at a constant temperature of 35 °C. The detectors were calibrated with a single poly(styrene) standard (Polymer Labs) and values of dn/dc (ml g−1) of 0.185 and 0.144 were used for poly(styrene) and poly(isoprene) respectively. Elemental micro-analysis (C, H, N, Br and F) was carried out on an Exeter Analytical, Inc. CE-440 Elemental Analyser. Melting points were determined on an Electrothermal 9100 capillary melting point apparatus.
Surface analysis
Thin films were prepared by spin coating onto clean silicon wafers using a Cammax PRS14E photoresist spinner to obtain film thicknesses of approximately 250 nm (measured by ellipsometry). Thin films comprised of blends of matrix polymer—poly(styrene) (PS) or poly(isoprene) (PI)—containing various concentrations of surface modifying additives of different molecular weight and different number of fluoroalkyl groups. The polymer blends were prepared by co-dissolving the required polymers in toluene to give 5% w/v solutions. Films were either analysed as unannealed—dried overnight under vacuum at room temperature to ensure complete evaporation of excess solvent—or annealed under vacuum at 150 °C for various annealing times. Static contact angles were measured at room temperature using a Ramé-Hart goniometer model 100. Angles were measured for annealed and unannealed films using water as the contact fluid, and all data quoted are the average of at least three and in most cases six individual contact angle measurements. A full statistical analysis of all the contact angle data has been carried out and the standard deviation for any individual data point varies from approximately 0.25 to 1.25 in a few extreme cases but in the majority of cases varies between 0.3 and 0.7—thus, an error bar representing a typical standard deviation of 0.5 has been added to Fig. 4 for illustrative purposes.
Rutherford backscattering analysis
The near-surface concentration of fluorocarbon was determined by Rutherford backscattering (RBS) analysis using an NEC 5SDH Pelletron accelerator. A 1.3 MeV 4He+ beam was brought onto the sample surface at 85° (i.e. 5° grazing incidence). Backscattered 4He+ ions were detected at 170° to the incident beam. Data were summed over 3 measurements of 1 µC 4He+ on different spots of the sample surface ensuring a quality of statistical results. This procedure has been previously shown to give reliable depth distribution data.47,51
Synthesis of endcapping agents
3,5-(Di-3-(perfluorooctyl)propyloxy)benzyl bromide (1).
Compound 1 was prepared in three steps according to the previously reported procedure.493-(Perfluorooctyl)propanol was converted to 3-(perfluorooctyl)propyl bromide (PFOPB) using carbon tetrabromide/triphenylphosphine (CBr4/PPh3) in dry THF/DCM. PFOPB was then added to 3,5-dihydroxybenzyl alcohol in dry acetone, in the presence of potassium carbonate and 18-crown-6 to yield 3,5-(di-3-(perfluorooctyl)propyloxy) benzyl alcoholvia a Williamson etherification reaction. Reaction with (CBr4/PPh3) converted the benzyl alcohol functionality to benzyl bromide to yield 1. 1H NMR (400 MHz, CDCl3, δ, ppm) 2.10 (m, 4H, CH2CH2CH2), 2.30 (m, 4H, CH2CF2), 4.02 (t, 4H, CH2O), 4.41 (s, 2H, CH2Br), 6.38 (t, 1H, ArH), 6.54 (d, 2H, ArH). Elemental analysis, calculated for C29H17BrF34O2: C, 31.01; H, 1.53; Br, 7.11; F, 57.50%. Found: C, 31.03; H, 1.50; Br, 6.97; F, 57.32%.
3,4,5-(Tri-3-(perfluorooctyl)propyloxy)benzyl bromide (2).
Compound 2 was prepared in 4 steps according to previously reported procedures.493-(Perfluorooctyl)propanol was converted to 3-(perfluorooctyl)propyl bromide (PFOPB) using carbon tetrabromide/triphenylphosphine (CBr4/PPh3) in dry THF/DCM. PFOPB was then added to methyl-3,4,5-trihydroxybenzoate in dry acetone, in the presence of potassium carbonate and 18-crown-6 to yield methyl-3,4,5-(tri-3-(perfluorooctyl)propyloxy)benzoate. Reaction with a suspension of fresh lithium aluminium hydride in dry THF yielded benzyl alcohol which in turn was treated with CBr4/PPh3 to yield 2. 1H NMR (400MHz, CDCl3, δ, ppm) 2.03 (m, 2H, CH2CH2CH2), 2.15 (m, 4H, CH2CH2CH2), 2.26–2.46 (m, 6H, CF2CH2), 4.01 (t, 2H, CH2O), 4.09 (t, 4H, CH2O), 4.44 (s, 2H, CH2Br), 6.63 (s, 2H, ArH). Elemental analysis, calculated for C40H22BrF51O3: C, 30.04; H, 1.39; Br, 5.00; F, 60.58%. Found: C, 29.91; H, 1.39; Br, 4.91; F, 54.15%.
Synthesis of polymer additives
All polymerisations were carried out by living anionic polymerisation using standard high vacuum techniques.
Poly(styrene) additives with 1CFgroup (PS1CF).
A typical PS1CF additive was prepared thus: benzene (100 ml) and styrene (7.29 g, 0.07 mol) were distilled, under vacuum, into the reaction apparatus. For a target molecular weight of 5000 g mol−1, s-butyllithium (BuLi) (1.04 ml of 1.4 M solution, 1.46 mmol) was added by injection via a rubber septum, resulting in the characteristic orange colour of polystyryllithium. The solution was stirred at room temperature for at least an hour (longer for higher molecular weights), after which time the reaction was terminated by the injection of 1H,1H,2H,2H-perfluorodecyldimethylchlorosilane (1.18 g, 2.19 mmol). The orange colour of the living polymer solution dissipated instantly. The polymer was recovered by precipitation into methanol, collected by filtration, washed with further methanol and dried to constant mass in vacuo. Yield >95%. Mn 5700 g mol−1, Mw 6000 g mol−1. 1H NMR (700 MHz, CDCl3, δ, ppm): −0.1 to −0.3 (6H, Si(CH3), 5.90–7.70 (m, 5H, ArH). A further three PS1CF additives were prepared by the same method—molecular weight analysis can be found in Table 1.
Table 1 Molecular weight and the extent of successful endcapping for a series of poly(styrene) (PS) and poly(isoprene) (PI) additives carry one (1CF), two (2CF) or three (3CF) C8F17 fluoroalkyl groups
| Additive |
Target Mw/g mol−1 |
M
n/g mol−1 |
M
w/g mol−1 |
% endcappinga |
|
Extent of endcapping calculated using 1H NMR.
|
|
PS1CF6K
|
5000 |
5700 |
6000 |
87 |
|
PS1CF11K
|
10000 |
11500 |
11800 |
74 |
|
PS1CF24K
|
20000 |
23600 |
24300 |
87 |
|
PS1CF40K
|
50000 |
39300 |
40600 |
96 |
|
PS2CF5K
|
5000 |
4900 |
5100 |
79 |
|
PS2CF9K
|
10000 |
9000 |
9400 |
63 |
|
PS2CF20K
|
20000 |
19500 |
20100 |
62 |
|
PS2CF44K
|
50000 |
43700 |
44800 |
60 |
|
PS3CF10K
|
10000 |
10100 |
10400 |
78 |
|
PS3CF28K
|
20000 |
27600 |
28100 |
43 |
|
PS3CF44K
|
50000 |
43500 |
44400 |
76 |
|
PI1CF5K
|
3000 |
5900 |
6400 |
80 |
|
PI1CF10K
|
6000 |
10500 |
10500 |
83 |
|
PI1CF17K
|
10000 |
17100 |
17700 |
82 |
|
PI1CF33K
|
20000 |
33300 |
33400 |
67 |
|
PI3CF11K
|
10000 |
10900 |
11200 |
50 |
Poly(styrene) additives with 2CFgroups (PS2CF).
A typical PS2CF additive was prepared thus: toluene (50 ml) and styrene (2.94 g, 0.03 mol) were distilled, under vacuum, into the reaction apparatus. For a target molecular weight of 5000 g mol−1, s-BuLi (0.42 ml of 1.4 M solution, 0.58 mmol) was injected via a rubber septum. The solution was stirred at room temperature for at least three hours (longer for higher molecular weights) before the addition of THF (25 ml) to the reaction flask by distillation, using a CO2/acetone bath to chill the receiving flask. Meanwhile, to a separate 25 ml round bottom flask fitted with a septum and sealed with a Young's tap was added 3,5-(di-3-(perfluorooctyl)propyloxy)benzyl bromide (0.80 g, 0.71 mmol, 1.2 equiv. w.r.t. lithium) which was dried under high vacuum for several hours/overnight. The endcapping agent was further purified azeotropically by the addition and subsequent removal (by distillation) of dry benzene. This process was repeated two more times and then the dried endcapping agent was dissolved in 10 ml purified THF. The THF solution of the endcapping agent was raised to atmospheric pressure with nitrogen gas, cooled in a CO2/acetone bath and then added by injection into the living polystyrene solution, at −78 °C, which resulted in the instant disappearance of the orange colour. The polymer was recovered by precipitation into an excess of methanol, collected by filtration, washed with further methanol and dried to constant mass in vacuo. Yield > 95%. Mn 4900 g mol−1, Mw 5100 g mol−1; 1H NMR (700 MHz, CDCl3, δ, ppm): 3.75–3.88 (4H, ArOCH2CH2) 5.90 (m, 2H, ArH), 6.20 (m, 1H, ArH) and 6.25–7.30 (m, 5H, ArH). A further three PS2CF additives were prepared by the same method—molecular weight analysis can be found in Table 1.
Poly(styrene) additives with 3CFgroups (PS3CF).
PS3CF additives were prepared in exactly the same fashion as that described above for PS2CF additives. Thus, toluene (50 ml) and styrene (3.00 g, 0.03 mol) were distilled, under vacuum, into the reaction apparatus. For a target molecular weight of 10000 g mol−1, s-BuLi (0.21 ml, 0.3 mmol) was injected via a rubber septum. The solution was stirred at room temperature for at least three hours (longer for higher molecular weights) before the addition of THF (25 ml) to the reaction flask by distillation using a CO2/acetone bath. Meanwhile, in a 25 ml round bottom flask (part of the same reactor and adjacent to the main reaction flask) 3,4,5-(tri-3-(perfluorooctyl)propyloxy)benzyl bromide (0.58 g, 0.36 mmol, 1.2 equiv w.r.t lithium), purified as described above, was dissolved in 10 ml purified THF. The THF solution of the endcapping agent was then cooled in a CO2/acetone bath and then decanted into the living polystyrene solution, at −78 °C, which resulted in the instant disappearance of the orange colour. The polymer was recovered by precipitation into an excess of methanol, collected by filtration, washed with further methanol and dried to constant mass in vacuo. Yield > 95%. Mn 10100 g mol−1, Mw 10400 g mol−1; 1H NMR (700 MHz, CDCl3, δ, ppm): 3.70–3.85 (4H, meta, ArOCH2CH2). 3.85–3.93 (2H, para, ArOCH2CH2). 5.80–5.96 (m, 2H, ArH), 6.25–7.35 (m, 5H, ArH). A further two PS3CF additives were prepared by the same method—molecular weight analysis can be found in Table 1.
Poly(isoprene) additives with 1CFgroup (PI1CF).
A typical PI1CF additive was prepared thus: benzene (100 ml) and isoprene (12.24 g, 0.18 mol), distilled from 0.1 ml n-BuLi immediately prior to use, were distilled under vacuum, into the reaction apparatus. For a target molecular weight of 3000 g mol−1, s-BuLi (2.91 ml of 1.4 M solution, 4.08 mmol) was injected via a rubber septum. The solution was stirred at room temperature overnight, after which time the reaction was terminated by the injection of 1H,1H,2H,2H-perfluorodecyldimethylchlorosilane (3.31 g, 6.12 mmol) and the reaction mixture was then left to stir overnight. The polymer was recovered by precipitation in excess methanol with a small amount of added antioxidant (BHT). The solvent was removed, the polymer redissolved in THF and reprecipitated into methanol, recovered and dried to constant mass in vacuo. Yield > 90%. Mn 4900 g mol−1, Mw 5100 g mol−1; 1H NMR (700 MHz, CDCl3, δ, ppm): −0.04 to −0.07 (6H, Si(CH3)2CH2), 0.70 (m, 3H, CH(CH3)CH2CH3, 0.90 (m, 3H, CH(CH3)CH2CH3). A further three PI1CF additives were prepared by the same method—molecular weight analysis can be found in Table 1.
Poly(isoprene) additives with 3CFgroups (PI3CF).
A single PI3CF additive was prepared as described above for PS2CF with some modifications. Thus, hexane (50 ml) and isoprene (5.00 g, 0.07 mol), distilled from 0.1 ml n-BuLi immediately prior to use, were distilled, under vacuum, into the reaction apparatus. For a target molecular weight of 10000 g mol−1, s-BuLi (0.36 ml, 0.5 mmol) was injected via a rubber septum and the solution was stirred at room temperature for two days. In order to remove any possible unreacted monomer from the reaction mixture, the reaction solvent was distilled from the reaction vessel and replaced with fresh, dry hexane. This process was repeated two further times, before dissolving the living polymer in 25 ml of fresh hexane. A small sample of polymer solution (for molecular weight/NMR analysis) was removed and quenched with nitrogen sparged methanol. Meanwhile, a mixture of diphenylethylene (DPE) and tetramethylethylene diamine (TMEDA) (1.5 and 2 mol equivalents respectively with respect to BuLi) was prepared in a 50 ml flask fitted with a rubber septum. The contents of the flask were frozen with liquid nitrogen and the air evacuated. Upon warming to room temperature the vessel was raised to atmospheric pressure with dry nitrogen and sec-BuLi solution added dropwise to titrate out any remaining impurities. It was concluded that the mixture was free of impurities when a persistent reddish colour of diphenylhexyllithium was observed. The mixture of the purified DPE/TMEDA was then injected into the living poly(isoprenyl)lithium and the mixture stirred at room temperature for 3 days, after which time the solution had developed an orange/red colour; a small sample was collected and quenched with nitrogen sparged methanol for analysis. To the DPE endcapped living polymer was added THF (25 ml) by distillation using a CO2/acetone bath. Meanwhile, in a separate 25 ml round bottom flask, 3,4,5-(tri-3-(perfluorooctyl)propyloxy)benzyl bromide (0.96 g, 0.6 mmol, 1.2 equiv. w.r.t. lithium), purified as described previously, was dissolved in 10 ml dry THF. The THF solution of the endcapping agent was then cooled in a CO2/acetone bath and then added to the living DPE endcapped poly(isoprene) solution at −78 °C. The orange colour of the living polymer slowly dissipated and after half an hour any remaining living chains were terminated with nitrogen sparged methanol. The polymer was recovered by precipitation in excess methanol containing a small amount of BHT. The solvent was removed, the polymer redissolved in THF and reprecipitated into methanol, recovered and dried to constant mass in vacuo. Yield > 90%. Mn 10900 g mol−1, Mw 112000 g mol−1; 1H NMR (700 MHz, CDCl3, δ, ppm): 3.28–3.39 (2H, ArCH2), 3.65–3.73 (m, 4H, meta, ArOCH2CH2), 3.87–3.95 (2H, para, ArOCH2CH2), 5.71–5.80 (m, 2H, ArH) (c), 4.92–5.27 (m, 1H, CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)CH2).
C(CH3)CH2).
Results and discussion
Synthesis of end functionalised polymer additives
We have previously described the synthesis of a limited number of similar polymer additives by atom transfer radical polymerisation49 and ring opening polymerisation47 in which the multifunctional end group was introduced through the use of a functionalised initiator. However, the sensitivity of living anionic polymerisation to functionality and impurities, limits the use of functionalised initiators and it was concluded that any methodology to synthesise end functionalised polymers of the type described herein, would be more productive if an endcapping strategy was adopted. To this end, a series of endcapping agents were used, with one, two or three C8F17 fluorocarbon (CF) groups (see Fig. 1). Endcapping agent 1CF (Fig. 1) is commercially available whereas 2CF and 3CF were synthesised according to previously reported procedures.49 Thus, a series of end functionalised polymers were prepared from both poly(styrene) (PS) and poly(isoprene) (PI). The synthesis of both PS and PI additives using 1CF endcapping agent follows well known and previously described methods.52PS1CF and PI1CF functionalised additives were prepared with molecular weights from 6000 to 40000 g mol−1 and molecular weights are reported in Table 1. In the case of PI1CF additives the constant error between the target and actual molecular weight was subsequently found to be due to the use of a bottle of initiator which had been contaminated and therefore the concentration of active lithium was less than the 1.4 M indicated. In general, the endcapping reaction resulted in greater than 80% end functionalisation of chains and that the endcapping reaction was not quantitative can almost certainly be attributed to the introduction of traces of impurities with the endcapping agent.
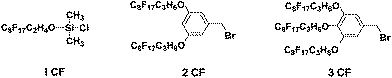 |
| | Fig. 1 Chemical structure of endcapping agents. | |
The synthesis of additives, end functionalised with 2CF and 3CF endcapping agents was a little more involved since the endcapping of polymers produced by living anionic polymerisation with alkyl halides (and derivatives) is often complicated by unwanted side reactions such as lithium–halogen exchange reactions and Wurtz coupling.53 In the case of endcapping polystyryllithium, the addition of a polar solvent such as THF to the reaction solvent and a reduced reaction temperature of −78 °C has been shown to have a positive impact on suppressing these side reactions and proved sufficient in this case.54,55 Using this approach, PS2CF and PS3CF additives were prepared with molecular weights in the range of 5000–44000 g mol−1 (see Table 1). Monomodal, narrow molecular weight distributions resulted (no chain coupling) and the extent of chain end functionalisation in some cases approached 80%. In other cases, though, the extent of endcapping was as low as 60–63% (PS2CF9K, PS2CF20K and PS2CF44K). These were some of the earliest endcapping experiments attempted and despite our best efforts to purify the endcapping agent, we suspect that impurities were probably introduced with the endcapping agent, resulting in termination of a proportion of the living chain ends. The endcapping agent cannot be distilled—it is a solid—so was purified by azeotropic drying with benzene. However, in order to transfer the ‘purified’ 2CF endcapping agent into the reaction flask it was necessary to do so by injection, which involved introducing nitrogen gas into the flask. We suspect that this may be the primary source of the impurities. This methodology was altered for the synthesis of the PS3CF additives with the use of a modified reactor. In this case the 3CF endcapping agent could be purified in the same way as the 2CF species but could be added directly to the main reaction flask without the need to introduce nitrogen gas. As a result it can be seen that PS3CF10K and PS3CF44K were successfully endcapped with improved extents of functionalisation of 78% and 76% respectively. However, to totally exclude all impurities from an endcapping reaction of a polymer produced by living anionic polymerisation with such a large and complex endcapping agent is extremely challenging! The relatively low extent of endcapping for PS3CF28K was due to a miscalculation of the quantity of endcapping agent necessary, with less than half the required amount added.
However, when the above described (successful) reaction conditions were applied to the endcapping of living poly(isoprene) with the benzyl bromide derivative, 3CF, the resulting polymer showed a very low degree of endcapping and a high degree of chain coupling. The propagating species at the chain end of polystyrene is a secondary (benzyl) carbanion whilst the propagating species of poly(isoprene), assuming high 1,4-microstructure, is a primary carbanion. Primary carbanions are more basic/nucleophilic than the styryl anion, and therefore more likely to undergo proton abstraction (leading to Wurtz coupling) during endcapping reactions, despite the addition of THF and reduced temperatures. It was therefore necessary to reduce the basicity/nucleophilicity of the propagating chain end prior to the addition of the endcapping agent. It has been shown that the most effective way of achieving this is to endcap the living polymer chain with diphenylethylene (DPE) prior to reaction with a benzyl halide.56DPE will react with a living carbanionic chain end as a monomer but due to its steric bulk will not undergo homopolymerisation.57 The result is the introduction of a single DPE unit whilst retaining the living nature of the polymeric chain end. Addition of the 3CF endcapping agent to the DPE endcapped poly(isoprene) did not result in the undesirable side reactions and no chain coupling was observed. However, the extent of endcapping was still modest (50%) in comparison to the analogous poly(styrene) additives. We suspect that this lower degree of endcapping was due to a combination of impurities and possibly, premature termination of the reaction. When endcapping poly(styrene), the orange colour of the living chain ends dissipated immediately, however the living DPE end group is less reactive and the reddish orange colour of the living DPE endcapped poly(isoprene) dissipated much more slowly. It appeared as if the colour had disappeared after 30 minutes but it is possible that some living chains were terminated (prematurely) by the addition of methanol rather than by the desired endcapping agent.
Analysis of surface properties
Analysis of surface properties was carried out on thin films of poly(styrene) and poly(isoprene) containing the described additives, to establish the relationship between the ability of the additives to modify the surface properties of the films and the molecular characteristics (molecular weight, number of CF groups and additive concentration) of the additives. Static contact angle measurements are a facile and convenient measure of the surface properties (hydrophobicity) of the films and contact angle analysis was carried out using water as the contact fluid. Measurements were initially carried out on ‘as-spun’ films and then repeated on films (of poly(styrene)) that had been annealed at 150 °C for a variety of times. Rutherford backscattering analysis was also carried out on examples of modified poly(isoprene) films to quantitatively analyse the effect of additive concentration and the number of fluoroalkyl groups on the near surface elemental composition of the modified thin films.
Contact angle analysis—poly(styrene) additives
Contact angle measurements were carried out on thin films of ‘bulk’ polystyrene (Mn = 90000 g mol−1, Mw/Mn = 1.10) containing various concentrations by weight of surface modifying additive. In each case the extent of endcapping was considered when calculating the amount required to create a particular blend composition. It can be seen from the data in Fig. 2 that some very clear relationships emerge between both the concentration of additive and the molecular parameters (molecular weight and number of CF groups) of the additive and surface properties. In all cases, increasing the concentration of additive leads to an initial sharp increase in contact angles at lower concentrations which is followed by a plateau in contact angle at higher concentrations of additive. That we observe significant changes to the surface properties compared to the pure unmodified polystyrene on smooth spin cast films implies strong surface segregation of the additives to the surface and that this segregation occurs during the spin coating process suggests high surface activity. The formation of a plateau or maximum contact angle might suggest that saturation of the surface with additive molecules has occurred, and the addition of further amounts of additive has little or no further impact upon the surface properties. However, the situation is somewhat more complex than that. That the fluorinated additives adsorb to the surface and generate an increase in contact angle is expected. The difference in surface energy between the two blend components allows the system to lower its overall free energy by having a surface enriched by the component with a lower surface energy—the fluorinated additive. As implied above, in reality, the distribution of a low surface energy component between bulk and surface, and therefore the surface composition, arises as a result of a series of interrelated phenomena and surface segregation of a low surface energy component can be hindered under specific conditions. For example, in a blend of block copolymer (with a low surface energy block) in a homopolymer matrix it has been demonstrated58 that the block copolymer can behave in a manner similar to that of small molecule surfactants in solution, forming large aggregates which adsorb (diffuse) very slowly to the surface. We believe that there are also strong similarities in the behavior of the fluorinated additives reported here and surfactants in solution.59 Moreover, the formation of a plateau in contact angles at higher concentrations of fluorinated additive is likely to be due to the formation of aggregates of polymer additives in the bulk rather than saturation of additives at the surface. The fluorinated head groups are relatively incompatible with the matrix poly(styrene) but at low concentrations the additives are molecularly dissolved—just as low concentrations of surfactants are in water. However, as the concentration of additives increases, there comes a point when the additives start to aggregate—just as surfactants form micelles! In the case of surfactants, the concentration at which this occurs is the critical micelle concentration (CMC); in the current work aggregation appears to occur above a critical aggregation concentration (CAC). In the case of surfactants, upon reaching CMC, the addition of further surfactant will serve only to increase the size and number of micelles and we suspect that above the CAC the addition of further additive merely results in an increase in the number/size of aggregates in the bulk phase. We have previously described similar behaviour59 for an analogous polymer additive with a molecular weight of 10000 g mol−1 carrying 2CFgroups made from deuterated polystyrene (dPS2CF10K). Small-angle neutron scattering (SANS) data showed that this additive forms aggregates at higher concentrations when in a matrix of poly(styrene). At concentrations of up to 12 wt% of dPS2CF10K, good agreement existed between experimental scattering data and a random phase approximation (RPA) simulation for linear polymer chains—implying molecularly dispersed individual chains. However, at higher concentrations i.e. 15 and 20 wt%, large deviations were found between experimental data and a similar simulation for discrete chains. When a modified simulation was carried out, in which it was assumed that the polymer additives were present as aggregates resembling star polymers, an excellent fit between experimental data and simulation was observed. As a result, the fitted experimental data for 15 and 20 wt% dPS2CF10K in hPS suggested that aggregates existed with an average of 6 or 7 dPS2CF10K polymer chains per aggregate. These data support the analogy that these fluorinated additives behave in a similar fashion to micelles in solution and it is certain that just as micelles are dynamic, there will be an equilibrium between free additive chains, molecularly dissolved in the bulk phase and aggregates (see Fig. 3). What is also certain is that the much larger aggregates will diffuse through the bulk at a much slower rate than the free chains and in effect only the free chains will be capable of surface segregation. There will also be an equilibrium between adsorbed chains at the surface and free chains in the bulk (in the presence of solvent or when above the glass transition temperature) and the distribution of additive chains between surface and bulk will be determined by both thermodynamic and kinetic factors, which in turn will be influenced by a number of parameters. The molecular weight of the additive will dictate the rate of diffusion through the bulk phase and the maximum packing density of additive chains at the surface. It is to be expected that low molecular weight additives will diffuse more quickly and pack more densely at the surface resulting in the higher contact angles observed for the lower molecular weight additives in Fig. 2. The number of CF groups will also play a huge role in this complex interplay of competing phenomena. Surface adsorption of the fluoroalkyl groups is thermodynamically favourable, leading to a reduction in the surface energy and once an additive is adsorbed, desorption is inhibited by the thermodynamic sticking energy—the thermodynamic glue which keeps the additive at the surface.59 The sticking energy is related to the amount of reduction in the surface energy which accompanies adsorption and therefore increasing the number of CF groups increases the sticking energy and further inhibits desorption. However, as the density of surface adsorbed chains increases we would expect an accompanying loss of conformational entropy and hence there is an entropic barrier to adsorption which competes with the sticking energy/reduction in surface energy. It is therefore expected that increasing the number of CF groups per additive chain, would result in a greater reduction in surface energy, driving the equilibrium distribution of chains in the system towards surface segregation and a concomitant increase in contact angles—as observed in Fig. 2. If we compare the observed contact angles for 3 additives of (nearly) identical molecular weight with 1, 2 and 3 CFgroups, PS1CF11K, PS2CF9K and PS3CF10K and at a constant concentration of 10 wt% we can see that the contact angle increases from 94° (1CF) to 99.5° (2CF) to 102.5° (3CF).
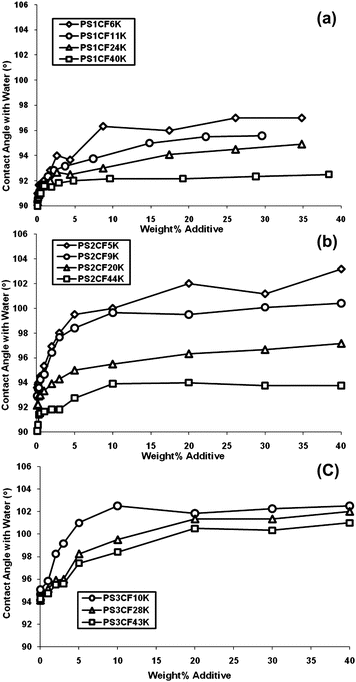 |
| | Fig. 2 Effect of molecular weight and the concentration of the additive upon static contact angle with water on thin (unannealed) films of polystyrene containing PS1CF (a), PS2CF (b) and PS3CF (c) additives. | |
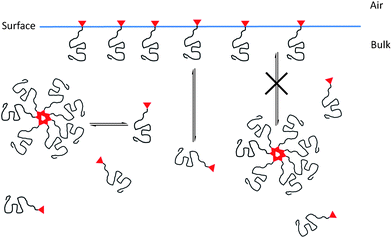 |
| | Fig. 3 Schematic demonstrating the proposed model of dynamic equilibria between free and aggregated additive chains in the bulk and between free additive chains in the bulk and at the surface. | |
Annealing of polystyrene thin films
It is highly unlikely that the distribution of additive chains in a thin film reaches equilibrium during the very short duration of the spin coating process. In order to establish the contact angle at equilibrium, films containing three concentrations of additive (from 0.5–5 wt%) were annealed at 150 °C (well above the glass transition temperature of poly(styrene)—104 °C) for a variety of annealing times. At this temperature the polymer chains are mobile and the films are able to reorganise, allowing enhanced surface segregation driven by a reduction in the surface energy. The data in Fig. 4 show the impact of annealing on films containing 0.5 wt% of the PS2CF additives and these data are typical. It can be seen that for all four molecular weights, annealing for up to three hours results in a significant increase in contact angle. Extended annealing beyond three hours results in further modest increases in contact angle and beyond seven hours at 150 °C there is little further change in surface properties—suggesting the system has reached equilibrium. What is remarkable is that annealing films containing very small quantities of additive (0.5%) can result in contact angles comparable to unannealed films containing substantially greater concentrations of additive. For example, annealing a film containing 0.5% PS2CF5K results in an equilibrium contact angle of approximately 98 degrees. To generate similar surface properties in an unannealed film requires approximately 6 times as much additive. Furthermore, in some cases annealing a film containing a relatively small quantity of additive can result in contact angles which exceed those achieved at all concentrations of additive in unannealed films. Thus comparing the data in Fig. 4 and 2b we can see that the maximum contact angle achieved in unannealed films containing up to 40 wt% of PS2CF44K is approximately 94 degrees whereas annealing a film containing as little as 0.5% of the same additive generates contact angles of almost 95 degrees and annealing a film containing 5% PS2CF44K results in a contact angle of nearly 97 degrees. The comparative differences in the contact angle under discussion are modest and the errors associated with contact angle analysis should not be ignored. To minimise the error and maximise our confidence in these data, each contact angle data point is the average of at least three and in most cases six individual contact angle measurements. In our experience the error in measuring contact angles is rarely higher than +/− one degree and statistical analysis of the contact angle data indicated that the standard deviation for any given data point was, in the majority of cases, between 0.3 and 0.7. To display error bars on each data point would unnecessarily clutter the figures, however an error bar representing a typical standard deviation of 0.5 has been added to Fig. 4 for illustrative purposes. Although the error in any given data point is in some cases comparable to the difference between datasets, we are confident that the trends observed are genuine and reproducible and Fig. 4 clearly shows that annealing films containing small quantities of additive has a dramatically beneficial impact on surface properties. The advantage of annealing films containing low concentrations of additive to allow them to reach equilibrium over simply adding greater quantities of additive is further demonstrated by the data in Table 2. In many cases, annealing films containing 5% (or less) results in contact angles in excess of those achieved by adding ever-increasing amounts of additive into a film which is subsequently not annealed. Most dramatic are the results for PS3CF10K where annealing a film containing 5% of this additive results in a contact angle of 107 degrees—5 degrees higher than that achieved in an unannealed film containing up to 40% additive, 17 degrees higher than unmodified poly(styrene) and approaching the surface properties of PTFE.49
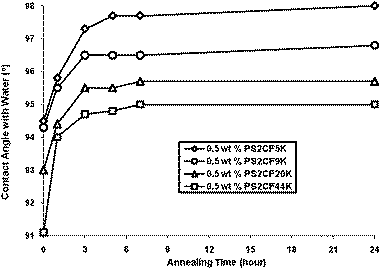 |
| | Fig. 4 Effect of annealing time (150 °C) on contact angle for PS2CF additives. | |
Table 2 Maximum contact angles achieved for a series of poly(styrene) films, unannealed (containing up to 40% additive) and annealed films containing varying concentrations of PS1CF, PS2CF and PS3CF additives
| Additive |
Maximum contact angle achieved |
| Unannealed filma |
Conc. of additive in film |
| 0.5% |
2% |
5% |
|
Maximum contact angle achieved for unannealed films containing up to 40% additive (see Fig. 2).
|
|
PS1CF6K
|
97.0 |
94.5 |
95.7 |
96.3 |
|
PS1CF11K
|
95.5 |
94.0 |
94.5 |
95.7 |
|
PS1CF24K
|
95.0 |
93.7 |
94.2 |
94.7 |
|
PS1CF40K
|
92.5 |
93.5 |
94.0 |
94.3 |
|
PS2CF5K
|
103.2 |
98.0 |
100.3 |
101.5 |
|
PS2CF9K
|
100.4 |
96.8 |
100.0 |
101.3 |
|
PS2CF20K
|
97.2 |
95.7 |
96.3 |
98.7 |
|
PS2CF44K
|
94.0 |
95.0 |
95.8 |
96.8 |
|
PS3CF10K
|
102.5 |
98.7 |
105.5 |
107.2 |
|
PS3CF28K
|
102.0 |
97.7 |
101.3 |
105 |
|
PS3CF43K
|
101.0 |
96.0 |
99.3 |
101.0 |
Contact angle analysis—poly(isoprene) additives
Contact angle measurements were also carried out on thin films of matrix poly(isoprene) (Mn = 88400 g mol−1, Mw/Mn = 1.02) containing various concentrations by weight of PI surface modifying additives. In each case the extent of endcapping was considered when calculating the amount required to create a particular blend composition. It can be seen from the data in Fig. 5 that similar relationships emerge between surface properties and the concentration, molecular weight and number of CF groups of the additive. In all cases increasing the concentration of additive leads to a concomitant increase in contact angles until a plateau in contact angles at higher concentrations of additive is observed. That we observe significant changes to the surface properties compared to unmodified poly(isoprene) implies strong surface segregation of the additives to the surface. The principal difference between the modified poly(styrene) and poly(isoprene) films is that poly(isoprene) is well above the glass transition temperature at room temperature and as such, in the case of poly(isoprene) the distribution of additive molecules between surface and bulk is able to reach equilibrium at room temperature. Thus the data in Fig. 5 were obtained from films which had been dried for 24 hours in vacuo at room temperature after spin coating. We are satisfied that that these data represent the equilibrium additive distribution since similar contact angle measurements taken over a 15 day period revealed no change in surface properties. The data in Fig. 5 also show a steady yet modest increase in the contact angle with the decreasing molecular weight of PI1CF additive but more dramatically shows the impact of the number of CF groups on surface properties. We can see that by comparing PI1CF10K and PI3CF11K, the additive with 3CFgroups has a contact angle which is approximately 10 degrees higher than that of an additive of nearly identical molecular weight with only 1CFgroup—these differences reinforce the dramatic impact of the effect of the number of CF groups upon surface properties seen for PS additives in this work and previously.49 Moreover, it is noteworthy that when additive PI3CF11K is present at concentrations above 2%, the contact angle exceeds that of PTFE (approximately 110 degrees49) by several degrees. One further important point to consider, when measuring contact angles on films of poly(isoprene), is that since poly(isoprene) is above it's glass transition temperature at room temperature, the film is able to reorganise in response to being in contact with water. Clearly the surface of both the unmodified and surface modified poly(isoprene) is highly hydrophobic and when placed in contact with water, the polymer chains in direct contact with the water droplet will attempt to minimise the interfacial tension and diffuse away from the surface. The concept of surface reorganisation in response to a new environment is well understood and has been previously described in the literature.60 The reorganisation would be expected to remove the CF groups from the interface and the contact angle decays over time. Indeed it is necessary to carry out the contact measurement immediately since the reorganisation of the films occurs in a matter of minutes and the contact angles decay steadily for a period of up to 45 minutes (the duration of the experiment). Whilst it might be argued that the decay in contact angle is due to evaporation of the solvent we don't believe this is the case even though the droplet of water was clearly evaporating. Evaporation of a droplet from a solid surface can result in a decay in contact angles, however it has been shown that when the liquid droplet is in contact with a non-wetting surface such as water on PTFE, evaporation results in a small (3–4 degrees) but rapid reduction in contact angle followed by a sustained period where evaporation occurs with a constant contact angle.61,62 In the present work the contact angle decayed at an almost constant rate over 45 minutes suggesting the reorganisation of the film surface. Intriguingly the rate of decay in contact angle also showed a strong dependence on the concentration of additive; these data will be the subject of a future publication.
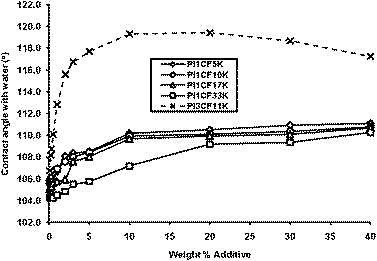 |
| | Fig. 5 Effect of the molecular weight and the concentration of the additive upon static contact angle with water on thin films of poly(isoprene) containing PI1CF and PI3CF additives. | |
Rutherford backscattering analysis
Although contact angle analysis is a very useful method of analysing surface properties, it is only an indirect method of analysis of the surface chemistry. In order to obtain quantitative analysis of the near surface elemental composition of the modified films, Rutherford backscattering (RBS) analysis was carried out on examples of modified poly(isoprene) films. This analysis enables an examination of the effect of additive molecular weight and the number of fluoroalkyl groups on the surface segregation and resulting surface composition of the modified thin films, confirming the relationship between these additive molecular parameters, surface adsorption and surface properties. The resulting data are therefore complimentary to the contact angle data. RBS analysis was carried out on modified poly(isoprene) films containing 5 wt% PI3CF11K, 20% PI3CF11K and 20% PI1CF10K and the data are shown in Fig. 6. The elemental markers in Fig. 6 indicate the maximum recoil energy of 4He+ ions from each element on the surface and since fluorine is the most massive element on the polymer surface, 4He+ recoils were detected at higher energy (570 keV) for fluorine, than recoils from other elements such as carbon and oxygen. The data in Fig. 6 for each film show a step in the detected counts for carbon in each film and the height of the carbon step can be used to normalize the data since the amount of carbon in the polymer film is known. It is therefore the size of the fluorine peak relative to the carbon signal that allows us to determine the amount of fluorine at the surface. At first glance, it would appear that of the three peaks arising from the presence of fluorine (at approximately 570 keV), the film containing 5 wt% PI3CF11K has the highest concentration of fluorine. However, although the fluorine peak for the film containing 5% PI3CF11K appears to be larger than that for the film containing 20% of the same additive it must be remembered that it is the size of this peak relative to the carbon signal at 350 keV that is important. RBS analysis can quantify fluorine adsorbed at the surface by providing a direct measurement in comparison to the bulk concentration. Simulations, in which the RBS spectrum of a thin layer of C8F17 on a thick film of poly(isoprene) was obtained, are also shown in Fig. 6 (solid lines). Simulations were carried out using the SIMNRA program63 and Rutherford scattering cross-sections. The thickness of the appropriate C8F17 layer was allowed to vary to provide the best fit to the relevant experimental data, and the surface concentration of fluorocarbon from this layer was determined. When the appropriate amount of fluorine due to the bulk concentration of additive was included in the simulation, excellent agreement between simulation and data in the energy range 300–600 keV was observed. Moreover, we can convert the apparent thickness of C8F17 surface layer in the SIMNRA simulation to a surface concentration of adsorbed fluorocarbon groups per unit area. The surface concentration of C8F17 fluorocarbon groups obtained for each film are shown in Table 3. It can be clearly seen from the data that the film containing 20 wt% PI3CF11K has the highest surface concentration of fluoroalkyl groups, which is followed closely by the film containing 5 wt% of the same additive. The film containing 20 wt% of an additive with nearly identical molecular weight but only 1CFgroup (PI1CF10K) has a significantly lower surface concentration of fluoroalkyl groups and confirms the dramatic impact that varying the number of CF groups can have. These data are consistent with the contact angle data shown in Fig. 5 where it can be seen that there is a significant difference in contact angle between PI3CF and PI1CF additives of the same molecular weight and rather more modest differences between the contact angle between films containing 5 wt% and 20 wt% of PI3CF11K. Furthermore, if one assumes that the C8F17 fluorocarbon groups lie parallel to the film surface, then each has an effective surface area of 0.65 nm2.47 In this orientation, the surface concentrations of additive molecule in each film correspond to a surface coverage of C8F17 groups of 13%, 81% and 88% for 20 wt% PI1CF10K, 5 wt% PI3CF11K and 20 wt% PI3CF11K respectively. At both concentrations of PI3CF we can see that the polymer film was covered with 80–90% fluorocarbon. It is clear from the RBS analysis results that the derived surface concentrations are consistent with the contact angle analysis and confirm that the blend surfaces are highly fluorinated and either increasing the number of fluoroalkyl groups or increasing the concentration of additive results in a greater density of fluorine at the surface.
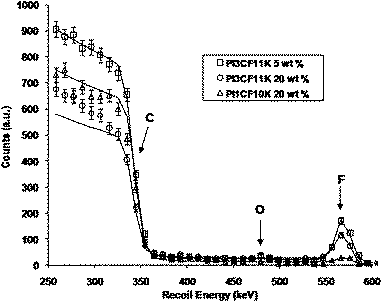 |
| | Fig. 6
RBS data and simulations for poly(isoprene) films modified with 5 wt% PI3CF11K, 20% PI3CF11K and 20% PI1CF10K. | |
Table 3 Calculated surface concentration of C8F17 groups in modified poly(isoprene) films containing 5 wt% PI3CF11K, 20% PI3CF11K and 20% PI1CF10K additive
| Sample |
Surface concentration C8F17 groups (mg m−2) |
Surface concentration C8F17 groups (molecules nm−2)a |
|
These values are derived from the previous values of unit molecules nm−2.
|
| 20 wt% PI1CF10K |
0.14 |
0.2 |
| 5 wt% PI3CF11K |
0.86 |
1.24 |
| 20 wt% PI3CF11K |
0.95 |
1.36 |
Conclusions
We describe here the development of synthetic methodologies to allow the synthesis, by living anionic polymerisation of a series of multi-end functionalised polymer additives, with 1, 2 and 3 fluoroalkyl groups, designed for the modification of polymer surfaces. Polymer additives were prepared from both poly(styrene) and poly(isoprene) with molecular weights in the range of 5000–50000 g mol−1. We have reported that increasing the additive concentration and/or number of CF groups results in enhanced hydrophobicity whereas increasing the molecular weight of additive had the opposite effect. The distribution of a low surface energy additive between the bulk and the surface of a film, and therefore the surface composition, arises as a result of a series of interrelated phenomena and we have argued that there are similarities in the behavior of the fluorinated additives studied in this work and surfactants in solution. Thus the formation of a plateau in contact angle (for unannealed films) at higher concentrations of fluorinated additive does not arise from saturation of the surface with additive but rather is due to the formation of aggregates (analogous to micelles) of polymer additives in the bulk above a critical aggregation concentration. This argument is further supported by annealing studies, which in all cases result in an increase in surface segregation as evidenced by an increase in contact angle after annealing. Annealing allows the system to reach equilibrium and the distribution of additives at equilibrium will be dependent upon both an equilibrium between free additive chains in the bulk and aggregated chains in the bulk and a second equilibrium between free chains at the surface and free chains in the bulk.
Poly(isoprene) has a glass transition temperature below room temperature and PI additives are able reach equilibrium without annealing at elevated temperatures. As a result of having a glass transition temperature below the room temperature, PI films are able to reorganise in the presence of a droplet of water and contact angles were seen to decay steadily over time. In view of the fact that contact angle data cannot propose any quantitative information on surface elemental composition, we have also carried out Rutherford backscattering analysis to quantify the concentration of fluorine near the surface of the film with respect to blend compositions that contain 1CF and 3CF poly(isoprene) additives. It is clear that when only 1CFgroup is present, the concentration of fluorine at the surface is low. However, for an equivalent concentration (20%) of additive and molecular weight of 3CF additive, the increase in functional groups results in approximately 90% coverage of fluoroalkyl groups at the surface. Moreover, even a far lower concentration (5 wt%) of the same additive can result in greater 80% surface coverage. RBS provides quantitative data which are complimentary to the contact angle data in so much that it confirms that increasing the number of fluoroalkyl groups or increasing the concentration results in a significant increase in the concentration of fluorine at the surface but that it is the number of fluoroalkyl groups that has the greatest impact on surface properties.
Acknowledgements
We acknowledge the financial support of the Malaysian Government and The University of Malaya for funding the PhD studies of Dr Muhamad-Sarih and support from the Department of Chemistry, Durham University.
References
- L. J. Ward, J. P. S. Badyal, A. J. Goodwin and P. J. Merlin, Polymer, 2005, 46, 3986 CAS.
- T. S. Cheng, H. T. Lin and M. J. Chuang, Mater. Lett., 2004, 58, 650 CrossRef CAS.
- E. Selli, G. Mazzone, C. Oliva, F. Martini, C. Riccardi, R. Barni, B. Macandalli and M. R. Massafra, J. Mater. Chem., 2001, 11, 1985 RSC.
- S. Sigurdsson and R. Shishoo, J. Appl. Polym. Sci., 1997, 66, 1591 CrossRef CAS.
- T. Kawase and H. Sawada, J. Adhes. Sci. Technol., 2002, 16, 1121 CrossRef CAS.
- T. Kawase, M. Yamane, T. Fuji and M. Minigawa, J. Adhes. Sci. Technol., 1997, 11, 1381 CrossRef CAS.
- S. Biltresse, D. Deschamps, T. Boxus and J. Marchand-Brynaert, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3510 CrossRef CAS.
- D. L. Schmidt, C. E. Coburn, B. M. DeKoren, G. E. Potter, G. F. Meyers and D. A. Fischer, Nature, 1994, 368, 39 CrossRef CAS.
- S. Saїdi, F. Guittard, C. Guimon and S. Géribaldi, Macromol. Chem. Phys., 2005, 206, 1098 CrossRef CAS.
- A. Mueller, T. Kowalewski and K. L. Wooley, Macromolecules, 1998, 31, 776 CrossRef CAS.
- D. J. Gan, A. Mueller and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3531 CrossRef CAS.
- C. S. Gudipati, C. M. Greenlief, J. A. Johnson, P. Prayongpan and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6193 CrossRef CAS.
- C. S. Gudipati, J. A. Finlay, J. A. Callow, M. E. Callow and K. L. Wooley, Langmuir, 2005, 21, 3044 CrossRef CAS.
- C. Cheng, K. L. Wooley and E. Khoshdel, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4754 CrossRef CAS.
- K. T. Powell, C. Cheng, K. L. Wooley, A. Singh and M. W. Urban, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4782 CrossRef CAS.
- D. R. Iyengar, S. M. Perutz, C. A. Dai, C. K. Ober and E. J. Kramer, Macromolecules, 1996, 29, 1229 CrossRef CAS.
- A. Boker, K. Reihs, J. G. Wang, R. Stadler and C. K. Ober, Macromolecules, 2000, 33, 1310 CrossRef.
- M. L. Xiang, X. F. Li, C. K. Ober, K. Char, J. Genzer, E. Siviniah, E. J. Kramer and D. A. Fisher, Macromolecules, 2000, 33, 6106 CrossRef CAS.
- J. Genzer, E. Siviniah, E. J. Kramer, J. G. Wang, M. L. Xiang, K. Char, C. K. Ober, R. A. Bubeck, D. A. Fischer, M. Graupe, R. Colorado, O. E. Shmakova and T. R. Lee, Macromolecules, 2000, 33, 6068 CrossRef CAS.
- T. Hayakawa, J. G. Wang, M. L. Xiang, X. F. Li, M. Ueda, C. K. Ober, J. Genzer, E. Siviniah, E. J. Kramer and D. A. Fischer, Macromolecules, 2000, 33, 8012 CrossRef CAS.
- L. Andruzzi, E. Chiellini, G. Galli, X. F. Li, S. H. Kang and C. K. Ober, J. Mater. Chem., 2002, 12, 1684 RSC.
- X. F. Li, L. Andruzzi, E. Chiellini, G. Galli, C. K. Ober, A. Hexemer, E. J. Kramer and D. A. Fisher, Macromolecules, 2002, 35, 8078 CrossRef CAS.
- J. P. Youngblood, L. Andruzzi, C. K. Ober, A. Hexemer, E. J. Kramer, J. A. Callow, J. A. Finlay and M. E. Callow, Biofouling, 2003, 19, 91 CrossRef CAS.
- A. Hexemer, E. Siviniah, E. J. Kramer, M. Xiang, X. Li, A. Fischer and C. K. Ober, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 411 CrossRef CAS.
- E. Martinelli, S. Menghetti, G. Galli, A. Glisenti, S. Krishnan, M. Y. Paik, C. K. Ober, D. M. Smilgies and D. A. Fischer, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 267 CrossRef CAS.
- G. Galli, A. Ragnoli, M. Bertolucci, C. K. Ober, E. J. Kramer and E. Chiellini, Macromol. Symp., 2004, 218, 303 CAS.
- G. Galli, E. Martinelli, E. Chiellini, C. K. Ober and A. Glisenti, Mol. Cryst. Liq. Cryst., 2005, 441, 211 CAS.
- S. Krishnan, N. Wang, C. K. Ober, J. A. Finlay, M. E. Callow, J. A. Callow, A. Hexemer, K. E. Sohn, E. J. Kramer and D. A. Fischer, Biomacromolecules, 2006, 7, 1449 CrossRef CAS.
- S. Krishnan, R. Ayothi, A. Hexemer, J. A. Finlay, K. E. Sohn, R. Perry, C. K. Ober, E. J. Kramer, M. E. Callow, J. A. Callow and D. A. Fischer, Langmuir, 2006, 22, 5075 CrossRef CAS.
- S. Krishnan, M. Y. Paik, C. K. Ober, E. Martinelli, G. Galli, K. E. Sohn, E. J. Kramer and D. A. Fischer, Macromolecules, 2010, 43, 4733 CrossRef CAS.
- T. F. Schaub, G. J. Kellogg, A. M. Mayes, R. Kulasekere, J. F. Ankner and H. Kaiser, Macromolecules, 1996, 29, 3982 CrossRef CAS.
- S. J. McLain, B. B. Sauer and L. E. Firment, Macromolecules, 1996, 29, 8211 CrossRef CAS.
- W.-K. Lee, I. Losito, J. A. Gardella, Jr and W. L. Hicks, Jr, Macromolecules, 2001, 34, 3000 CrossRef CAS.
- Z. Su, D. Wu, S. L. Hsu and T. J. McCarthy, Macromolecules, 1997, 30, 840 CrossRef CAS.
- M. O. Hunt, A. M. Belu, R. W. Linton and J. M. DeSimone, Macromolecules, 1993, 26, 4854 CrossRef.
- S. Affrossman, M. Hartshorne, T. Kiff, R. A. Pethrick and R. W. Richards, Macromolecules, 1994, 27, 1588 CrossRef CAS.
- C. Yuan, M. Ouyang and J. T. Koberstein, Macromolecules, 1999, 32, 2329 CrossRef CAS.
- K. Tanaka, D. Kawaguchi, Y. Yokoe, T. Kajiyama, A. Takahara and S. Tasaki, Polymer, 2003, 44, 4171 CrossRef CAS.
- P. A. O'Rourke Muisener, S. K. Kumar and J. T. Koberstein, Macromolecules, 2003, 36, 771 CrossRef CAS.
- P. A. V. O'Rourke Muisener, C. A. Jalbert, C. Yuan, J. Baetzold, R. Mason, D. Wong, Y. J. Kim and J. T. Koberstein, Macromolecules, 2003, 36, 2956 CrossRef.
- K. Sugiyama, A. Hirao and S. Nakahama, Macromol. Chem. Phys., 1996, 197, 3149 CrossRef CAS.
- A. Hirao, G. Koide and K. Sugiyama, Macromolecules, 2002, 35, 7642 CrossRef CAS.
- A. Hirao, K. Sugiyama and H. Yokoyama, Prog. Polym. Sci., 2007, 32, 1393 CrossRef CAS.
- A. A. El-Shehawy, H. Yokoyama, K. Sugiyama and A. Hirao, Macromolecules, 2005, 38, 8285 CrossRef CAS.
- H. Li, Y. M. Zhang, H. Zhang, M. Z. Xue and Y. G. Liu, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3853 CrossRef CAS.
- L. R. Hutchings, A. P. Narrianen, R. L. Thompson, N. Clarke and L. Ansari, Polym. Int., 2008, 57, 163 CrossRef CAS.
- L. R. Hutchings, A. P. Narrainen, S. M. Eggleston, N. Clarke and R. L. Thompson, Polymer, 2006, 47, 8116 CrossRef CAS.
- A. L. Narrainen, L. R. Hutchings, I. A. Ansari, N. Clarke and R. L. Thompson, Soft Matter, 2006, 2, 126 RSC.
- A. P. Narrainen, L. R. Hutchings, I. Ansari, R. L. Thompson and N. Clarke, Macromolecules, 2007, 40, 1969 CrossRef CAS.
- R. L. Thompson, A. P. Narrainen, S. M. Eggleston, I. A. Ansari, L. R. Hutchings and N. Clarke, J. Appl. Polym. Sci., 2007, 105, 623 CrossRef CAS.
- F. T. Kiff, R. W. Richards and R. L. Thompson, Langmuir, 2004, 20, 4465 CrossRef CAS.
- L. R. Hutchings, R. W. Richards, R. L. Thompson, D. G. Bucknall and A. S. Clough, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 5, 451 Search PubMed.
-
H. L. Hseih and R. P. Quirk, Anionic Polymerization, Principles and Practical Applications, Marcel Dekker, Inc, New York, 1996 Search PubMed.
- M. Gauthier and M. Moller, Macromolecules, 1991, 24, 4548 CrossRef CAS.
- R. A. Kee and M. Gauthier, Macromolecules, 1999, 32, 6478 CrossRef CAS.
- C. M. Fernyhough, R. N. Young, A. J. Ryan and L. R. Hutchings, Polymer, 2006, 47, 3455 CrossRef CAS.
- L. R. Hutchings, J. M. Dodds and S. J. Roberts-Bleming, Macromolecules, 2005, 38, 5970 CrossRef CAS.
- Y. Lyatskaya, D. Gersappe, N. A. Gross and A. C. Balazs, J. Phys. Chem., 1996, 100, 1449 CrossRef CAS.
- I. A. Ansari, N. Clarke, L. R. Hutchings, A. Pillay-Narrainen, A. E. Terry, R. L. Thompson and J. R. P. Webster, Langmuir, 2007, 23, 4405 CrossRef CAS.
- J. T. Koberstein, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 2942 CrossRef CAS.
- T. Furuta, A. Nakajima, M. Sakai, T. Isobe, Y. Kameshima and K. Okada, Langmuir, 2009, 25, 5417 CrossRef CAS.
- G. McHale, S. M. Rowan, M. I. Newton and M. K. Banerjee, J. Phys. Chem. B, 1998, 102, 1964 CrossRef CAS.
-
M. Mayer, SIMNRA User's Guide, 9/113, Max-Planck-Institut für Plasmaphysik, Garching, 1997 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.