Mesh-size control and functionalization of reorganizable chemical gels by monomer insertion into their cross-linking points
Yoshifumi
Amamoto
a,
Moriya
Kikuchi
ab,
Hiroyasu
Masunaga
c,
Hiroki
Ogawa
c,
Sono
Sasaki
c,
Hideyuki
Otsuka
*ad and
Atsushi
Takahara
*abd
aInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: otsuka@ms.ifoc.kyushu-u.ac.jp; takahara@cstf.kyushu-u.ac.jp
bERATO, Japan Science and Technology Agency, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
cJapan Synchrotron Radiation Research Institute/SPring-8, 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
dInternational Research Center for Molecular Systems, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
First published on 27th January 2011
Abstract
Mesh-size control of network structures of chemical gels, i.e. mesh expansion by insertion of styrene derivatives and mesh shrinking by insertion of divinylbenzene in the gels, was carried out. Chemical gels with alkoxyamine at their cross-linking points were synthesized by radical copolymerization of methyl methacrylate and divinyl monomer with alkoxyamine units. The monomers were inserted by heating the gels with each monomer separately, and the network size was evaluated by small-angle X-ray scattering (SAXS) measurements. Since a living polymerization procedure was adopted for the reactions, network mesh sizes were controlled precisely by controlling the reaction time and selecting an appropriate monomer. In addition, gel properties such as swelling degree and phase separation could also be controlled by employing functional monomers such as hydrophilic and fluorinated monomers. These reorganizable chemical gels combine the advantages of high stability of chemical gels with ease of structural changeability of physical gels.
1 Introduction
Polymer gels have attracted considerable attention, because they provide several different functionalities owing to their cross-linking structures, such as incorporation of compounds, volume phase transitions,1 swelling,2 and self-healing.3 Thus far, several functional polymer gels have been reported, including topological gels,4 nanocomposite gels,5 double-network gels,6polymer gels with peristaltic motion,7 and tetra-PEG gel.8 Generally, polymer gels are classified according to their cross-linking structure as chemical gels or physical gels, with cross-linking points that consist of covalent bonds and non-covalent interactions, respectively.The main advantages of chemical gels are their good mechanical properties and high stability; however, one drawback of these gels is difficulty in changing the gel network structure after its preparation. Because the polymer chains of these gels are connected covalently, it is topologically difficult to change their structures. However, if a reversible covalent bond9–12 would be introduced into the cross-linking points, both the stability and the ability to change the network structure would improve.13 Even though some bonds are cleaved temporarily, the other non-cleaved bonds retain their stabilities and high mechanical strengths. Some examples of functional gels with reversible covalent bonds have been reported previously; sol–gel transition,14–17 photo-induced plasticity,18 self-healing materials,14,19 and so on have been achieved.
We have also developed chemical gels with reversible covalent bonds, ‘reorganizable chemical gels’; they contain alkoxyamine (C–ON) units.20 Although some alkoxyamine compounds derived from styryl radicals and nitroxide radicals, such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), behave as typical covalent compounds, they polymerize styrene and acrylate monomers under heating conditions, well known as nitroxide-mediated radical polymerization (NMRP),21–23 and they promote crossover reactions between radicals.22 Thus far, we have achieved sol–gel transitions20,24 as well as structural transformations between block copolymers and nanogels.25–29
In a previous paper, we reported insertion of styrene monomers into cross-linking points of polystyrene backbone gels with alkoxyamine by the NMRP process.20 Such insertion has potential to enable precise control of network size and functionalization of the cross-linked polymers. In this study, reorganizable chemical gels with poly(methyl methacrylate) backbones and alkoxyamine at their cross-linking points were designed, and precise mesh-expanding reactions viainsertion of mono-functional monomers and mesh-shrinking reactions viainsertion of bi-functional monomers were achieved by NMRP, as shown in Fig. 1.
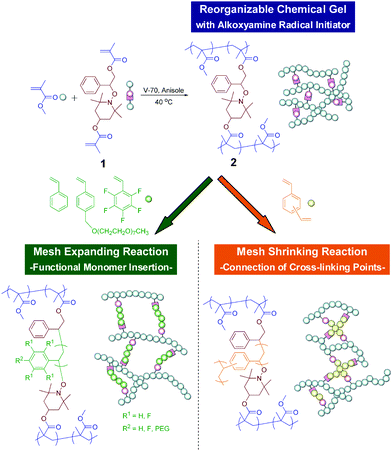 | ||
| Fig. 1 Chemical structures and models of reorganizable chemical gels, and their functional monomer insertion and mesh shrinking reactions. | ||
2 Experimental
2.1 Materials
4-Hydroxy-1-((2′-hydroxy-1′-phenylethyl)oxy)-2,2,6,6-tetramethylpiperidine (alkoxyamine diol)30 was prepared and purified as previously reported. Methacryloyl chloride was purchased from Tokyo Chemical Industry, and used without further purification. Triethylamine (99%), methyl methacrylate (MMA, 99%), anisole (99%), styrene (99%), and divinyl benzene (DVB) were purchased from Wako Pure Chemical Industries, and purified by distillation under reduced pressure over calcium hydride. 2,2′-Azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70, 95%) was purchased from Wako Pure Chemical Industries and used without further purification. 2,3,4,5,6-Pentafluorostyrene (PFSt) was purchased from Tokyo Chemical Industry, purified by distillation under reduced pressure over calcium hydride. Tetrahydrofuran (THF, 99.5%) was purchased from Wako Pure Chemical Industries and purified with a Glass Contour solvent purification system. Polyethylene glycol monomethyl ether (PEG, nPEG = 7) was purchased from Alfa Aesar, and used without further purification. 4-(Chloromethyl)styrene was kindly supplied by AGC Seimi Chemical Co., Ltd. and used without further purification.2.2 Measurements
1H (300 MHz) NMR spectroscopic measurements were carried out at 25 °C with a JEOL JNM-AL300 spectrometer using tetramethylsilane (TMS) as an internal standard in chloroform-d (CDCl3).SAXS measurements. SAXS measurements were carried out at the BL40B2 beamline of SPring-8 using an incident X-ray with wavelength λ = 0.150 nm. Scattered X-rays were detected using a 300 mm × 300 mm imaging plate with a resolution of 0.1 mm/pixel and 2187 mm sample-to-detector distance calibrated by the average of eleven peaks of collagen. The measured gels were swollen in THF and contained in 2 mm glass capillaries. The scattering intensity of the polymers (ΔI(q)) was calculated by subtracting the scattering intensity of the solventIsolv(q) from that of the solution Isoln(q) adjusted through transmittance (Tsolv and Tsoln), as a following equation: ΔI(q) = Isoln(q)/Tsoln − Isolv(q)/Tsolv
Swelling degree measurements. The swelling degrees were estimated by following procedures: swelling weight (Wswelling) was measured after swelling cross-linked polymers into water for 72 h. After that, the swollen gel was dried under vacuum for 24 h, and the gel weight (Wgel) was measured. The swelling degree was calculated using following equation [= (Wswelling − Wgel)/Wgel (wt/wt)].
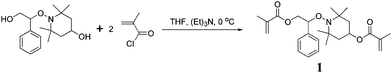
2.3 4-(Methacryloyloxy)-1-((2′-(methacryloyloxy)-1′-phenyl ethyl)oxy)-2,2,6,6-tetramethylpiperidine (1).
Alkoxyamine diol (2.93 g, 10 mmol) was charged into a round-bottom flask and dried under vacuum. After argon replacement, THF (40 mL) and triethylamine (4.18 mL, 30 mmol) were charged into the round-bottom flask, and then methacryloyl chloride (2.93 mL, 30 mmol) was added dropwise via syringe in an ice bath. After stirring for 12 h, methanol (5 drops) was added, and the solvent was removed under vacuum. Water and dichloromethane were added, then the aqueous layer was washed with dichloromethane, and the combined organic layer was evaporated. The crude product was purified by silica-gel chromatography eluting with hexane–chloroform (1/2, v/v) and dried in vacuo to give divinyl monomer 1 as a colorless transparent oil (1.96 g, 46% yield). 1H NMR: δ (ppm) 0.71 (s, 3H, CH3), 1.15 (s, 3H, CH3), 1.31 (s, 3H, CH3), 1.39 (s, 3H, CH3), 1.50 (d, J = 12 Hz, 1H, CH2), 1.59 (d, J = 10 Hz, 1H, CH2), 1.66 (d, J = 12 Hz, 1H, CH2), 1.79 (d, J = 13 Hz, 1H, CH2), 1.86 (s, 3H, CH3), 1.91 (s, 3H, CH3), 4.34 (dd, J = 11 Hz, 6 Hz, 1H, CH2), 4.65 (dd, J = 11 Hz, 6 Hz, 1H, CH2), 4.98 (t, J = 6 Hz, 1H, CH), 5.06 (m, 1H, CH), 5.49 (s, 1H, CH2), 5.52 (s, 1H, CH2), 6.00 (s, 1H, CH2), 6.06 (s, 1H, CH2), 7.27–7.33 (m, 5H, aromatic). 13C NMR: δ (ppm) 16.16, 19.01, 31.90, 42.51, 42.60, 58.31, 58.54, 64.23, 64.73, 82.04, 123.14, 123.57, 125.61, 125.73, 125.99, 134.04, 134.52, 138.14, 164.91, 165.02. FT-IR (NaCl, cm−1): 2978 (C–H), 1711 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1638 (CC), 1171, 907, 650. HRMS exact mass calculated for [M + 1]+C25H36NO5 430.2593, found 430.2595.
O), 1638 (CC), 1171, 907, 650. HRMS exact mass calculated for [M + 1]+C25H36NO5 430.2593, found 430.2595.
2.4 Cross-linked polymer with alkoxyamine units (2)
In a typical run (about 2a), divinyl monomer 1 (0.859 g, 2 mmol), MMA (1.93 mL, 18 mmol), anisole (4.28 mL), and V-70 (31 mg, 0.10 mmol) were charged into a test tube fitted with a rubber septum, and argon replacement was performed via bubbling. Then the test tube was immersed in an oil bath thermostatted at 40 °C under argon atmosphere. After 100 h, the cross-linked polymer was purified by Soxhlet extraction with dichloromethane, and the remaining solid was collected and dried under vacuum to give a colorless solid. (2.67 g, 99% yield).2.5 p-(Polyethylene glycol) styrene (PEGSt)
PEGSt was synthesized from 4-(chloromethyl)styrene by reference to a previous report using a polydisperse PEG (Mn = 350).31 After a purification by a silica-gel column chromatography (ethyl acetate–hexane = 4/1, v/v), a mixture of three molecular weight PEGSt (n(PEG) = 6–8) was obtained.2.6 Polymerization of styrene derivatives from cross-linked polymers with alkoxyamine at cross-linking points
In a typical run, cross-linked polymer 2b (100 mg), styrene (246 μL), and anisole (246 μL) were charged into a glass tube. The solution was degassed by seven freeze-pump-thaw cycles. Subsequently, the glass tube was sealed off under vacuum, and heated at 100 °C for 8 h. (138 mg, conv. 27%). The conversion of styrene derivatives were estimated by NMR measurements using anisole as an internal standard.3 Results and discussion
3.1 Preparation and characterization of reorganizable chemical gels
Polymer gels with alkoxyamine units at the cross-linking points were prepared by two-step syntheses: preparation of a divinyl monomer, and radical copolymerization of MMA and the divinyl monomers. The divinyl monomer (1) was synthesized by a condensation reaction between alkoxyamine diol and methacryloyl chloride, as indicated in Scheme 1, and the structure was confirmed by NMR, IR and mass measurements. Subsequently, the polymer gels with alkoxyamine units (2) were prepared by radical copolymerization of MMA and 1 at three comonomer ratios ([MMA]:[1] = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 19:1, and 49:1) using V-70 as a radical initiator in anisole at 40 °C for 100 h, as shown in Fig. 1. In all cases, the polymer gels formed after several tens of minutes or hours.
:1, 19:1, and 49:1) using V-70 as a radical initiator in anisole at 40 °C for 100 h, as shown in Fig. 1. In all cases, the polymer gels formed after several tens of minutes or hours.
 | ||
| Scheme 1 Synthesis of divinyl monomer 1 by a condensation reaction of alkoxyamine diol and methacryloyl chloride. | ||
Table 1 summarizes the conversions and yields of polymer gel syntheses under three copolymerization conditions. High conversions were obtained in the three cases, indicating that hardly any unreacted vinyl monomers remained in the gels. The yields also show high values, whereas in the case of 2c a comparatively low value (85%) was confirmed. It is conceivable that linear polymers and polymers with fewer cross-linking points were removed at the purification stage, because of lower cross-linking density of 2c. Fig. 2a shows photographs of polymer gels 2a, and clear and lens-like structures in the glass tubes were observed. This is because the main chains of the gels are poly(methacrylate)s, which show high transparency even in the gel state.
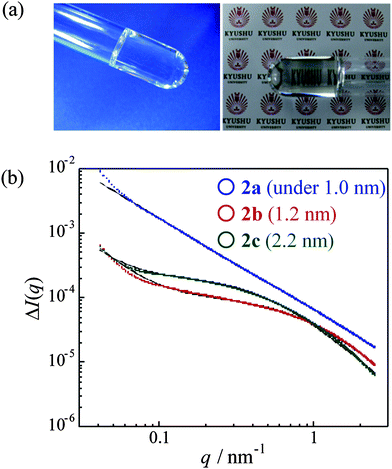 | ||
| Fig. 2 (a) Photographs of cross-linked polymers 2a, and (b) SAXS profiles of cross-linked polymers 2, their fitting curves (black), and each evaluated correlation length (ξ). | ||
The cross-linking states of three types of the gels differing in number of cross-linking points were investigated by small-angle X-ray scattering (SAXS) measurements using synchrotron radiation at BL40B2 in SPring-8. In static scatterings, Ornstein-Zernike (OZ) equations32 are generally used to evaluate network structures, and their correlation length ξ can be treated as the mesh size. Additionally, heterogeneous structures for cross-linking states should be considered,33 especially in chemical gels. Fig. 2b shows the SAXS profiles for 2a (blue), 2b (red), 2c (green), and their fitting curves (black), which is the sum of the term from the contribution of fractal structures to represent a heterogeneous structure (in the lower q range) and an OZ equation (in the higher q range),34 as follows:
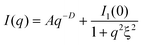 | (1) |
3.2 Mesh expanding reaction by polymerization of monofunctional styrene derivatives
The mesh-expanding reaction of the chemical gels was carried out by polymerizing mono-functional styrene derivatives (styrene, PEGSt, and PFSt) from alkoxyamine units at the cross-linking points. In the case of polymer gel 2a with the highest cross-linking density, the polymerization of styrene derivatives hardly proceeded, because the dissociation of the alkoxyamine units in high cross-linked polymer gels was negligible. In the case of 2c, the polymerization rate was slow compared with 2b, because the concentration of the alkoxyamine units in the gel was lower. With this in mind, polymer gel 2b was utilized in subsequent reactions. The reactions were carried out by heating a mixture of polymer gel 2b and each styrene derivative (50 mol equiv/alkoxyamine in 2b) in anisole ([monomer]/[anisole] = 1/1 (v/v)). In consideration of monomer reactivity, styrene was polymerized at 100 °C, and PEGSt and PFSt were polymerized at 110 °C. Before heating, the gels were not completely swollen in the solution. However, with increasing reaction time, the gel volume gradually increased and the surrounding solution disappeared, indicating that the monomers were consumed and incorporated into the cross-linking points. Indeed, the strength of the reacted gels before heating differed significantly from that after heating, and the gels gradually softened as the reaction proceeded.When the radical concentration due to dissociation of alkoxyamine units is constant, the polymerization rate in a first-order reaction should only depend on the monomer concentration, and ln([M]0/[M]) values should increase linearly with reaction time. Fig. 3a shows the kinetic plots (ln[M]0/[M] versus reaction time) in the cases of styrene and PEGSt. Here, [M]0 and [M] are molar concentrations of monomers or styrene derivatives before and after the reactions, respectively, in each reaction time, as determined by NMR measurements of extracted solutions using anisole as an internal standard. In both cases, the ln[M]0/[M] values increased linearly with reaction time, indicating that the monomers were consumed at the same polymerization rate constants. This implies that polymerization proceeded with constant radical concentrations. In other words, the alkoxyamine units are living radical polymerization initiators, and these data confirms that the styrene derivatives were inserted into the cross-linking points of the chemical gels via the NMRP process.
![Time dependence of (a) ln([M]0/[M]), and (b) correlation length ξ after heating the gels 2b with styrene at 100 °C (circles), and PEGSt at 110 °C (triangles) with anisole ([monomer]/[anisole] = 1/1 (v/v)).](/image/article/2011/PY/c0py00304b/c0py00304b-f3.gif) | ||
| Fig. 3 Time dependence of (a) ln([M]0/[M]), and (b) correlation length ξ after heating the gels 2b with styrene at 100 °C (circles), and PEGSt at 110 °C (triangles) with anisole ([monomer]/[anisole] = 1/1 (v/v)). | ||
The network structures during the polymerization of styrene derivatives were characterized by SAXS measurements, and the correlation length values (ξ) were evaluated by OZ functions, as seen in Fig. 3b. In the both the cases of styrene and PEGSt, ξ increased with increasing reaction times, and the ξ values reached from 1.17 nm to 1.75 nm (styrene) and 1.50 nm (PEGSt) after 8 h. This result indicates an increase in the mesh size, because the monomers were inserted into the cross-linking points, as shown in Fig. 1 (left). The characteristic feature of this reaction system is that the mesh sizes are controlled by the reaction time; this is attributed to the fact that alkoxyamine units were used for the living polymerization of styrene derivatives.
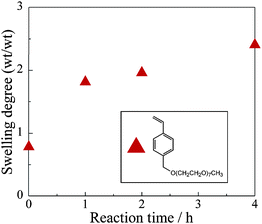
In the case of insertion of functional monomers such as PEGSt, the gel properties were expected to undergo notable changes, and this was investigated on the basis of the swelling behaviour of the gel in water. Fig. 4 shows the swelling degree (= (Wswelling − Wgel)/Wgel (wt/wt)) for water after heating 2b with PEGSt for several reaction times. The swelling degree against water increased with increasing reaction time. This is because the hydrophilic PEGSt was polymerized at the cross-linking points, and subsequently, the amount of PEG in the gel increased. As is the case with the mesh size, the swelling degree can also be controlled easily by controlling the reaction time.
In X-ray measurements, differences of electron densities enable the discussion of polymer structures such as phase separations, and fluorinated monomer with high electron density was used as a marker for SAXS measurements. PFSt was polymerized by heating 2b in anisole at 110 °C. Fig. 5 shows SAXS profiles of polymers in THF before and after polymerization of PFSt for 4 h and 8 h. With increasing reaction times, peaks that could not be represented by eqn (1) were confirmed, as indicated by arrows. It was conceivable that the phase separation states were shown, because the electron density of PFSt at the cross-linking points was higher than that of PMMA in the main chain. The sizes of phase separation (2π/q) were estimated to be about 30 nm, which might correspond to the size of higher-cross-linking parts in the chemical gels, because common chemical gels are heterogeneous consisting of regions with varying cross-linking densities.
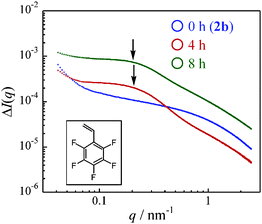 | ||
| Fig. 5 SAXS profiles of polymer gels after heating 2b with pentafluorostyrene at 110 °C in anisole for 0 h (blue), 4 h (red), and 8 h (green). | ||
3.3 Mesh shrinking reaction
Although we have carried out mesh-expanding reactions in this study and in a previous study,20 the reorganizable chemical gels considered here also have potential for undergoing a mesh-shrinking reaction. Bi-functional monomers could be proposed as sensible candidates for this purpose, because the two vinyl groups connect cross-linking points and form smaller mesh structures, as shown in Fig. 1 (right). The mesh-shrinking reaction was carried out by heating 2b with divinylbenzene (DVB, 50 mol equiv/alkoxyamine in 2b) in anisole ([DVB]/[anisole] = 1/1 (v/v)) at 100 °C. Similar to mesh-expanding reactions, in the mesh-shrinking reaction as well, the gels were not completely swollen before heating, and the surrounding solution gradually disappeared through the reaction. However, the gel state in this case was quite different from that in the mesh-expanding reaction; the gel assumed a rigid state with increasing reaction time, which indicates the increase in cross-linking density.Fig. 6a shows the kinetic plot (ln[M]0/[M] versus reaction time) in the mesh-shrinking reaction. The molar concentration of DVB ([M]) in each reaction time was estimated by the consumption of the vinyl group in the NMR measurement and not by the conversion of the monomer. The plot was almost linear until 4 h, and deviated from the linearity after 8 h. This is because the gel became more and more dense, causing the insertion of monomers into alkoxyamines to be more difficult. However, the reaction did not stop but continued to proceed slowly even after 8 h. In other words, the NMRP proceeded almost without any side reaction of alkoxyamine units such as carbon-carbon coupling until 4 h.
![Time dependence of (a) ln([M]0/[M]), and (b) ξ after heating 2b with DVB swollen in anisole ([DVB]/[anisole] = 1/1 (v/v)) at 110 °C. The ξ values were estimated by fitting OZ functions to scattering profiles. (c) Illustrations of “inter” and “intra” cross-linking reactions.](/image/article/2011/PY/c0py00304b/c0py00304b-f6.gif) | ||
| Fig. 6 Time dependence of (a) ln([M]0/[M]), and (b) ξ after heating 2b with DVB swollen in anisole ([DVB]/[anisole] = 1/1 (v/v)) at 110 °C. The ξ values were estimated by fitting OZ functions to scattering profiles. (c) Illustrations of “inter” and “intra” cross-linking reactions. | ||
Variations in the mesh size during polymerization of DVB were investigated by SAXS measurements, and the correlation length (ξ) was estimated by fitting OZ functions to the scattering profiles. Fig. 6b shows the time dependence of ξ under the same polymerization conditions as those in the above-discussed reactions. The ξ values decreased with increasing reaction time, from 1.17 nm to 0.70 nm after 8 h. This result indicates that the mesh sizes decreased and the mesh-shrinking reaction was completed successfully. Similar to the mesh-expanding reaction, in the mesh-shrinking reaction as well, the mesh size was controlled by the reaction time, because the mesh-shrinking reaction was based on the NMRP process. These reactions can be explained by two possible mechanisms: connection of “inter” cross-linking points and formation of PDVB at “intra” cross-linking points as illustrated in Fig. 6c. We consider that the mesh-shrinking reaction occurred by an intricate combination of these two mechanisms.
4 Conclusions
In summary, we demonstrated the mesh expanding, shrinking reactions, and functional monomer insertion for reorganizable chemical gels. Chemical gels with alkoxyamine units were prepared via free-radical copolymerization of MMA and bifunctional monomers with alkoxyamine units, and transparent gels were quantitatively obtained. The mesh expanding and shrinking reactions of the gels were achieved by heating the gels with monofunctional monomers (styrene, PEGSt, and PFSt) and bifunctional monomer (DVB), respectively. From the aspect of kinetic behavior, the reactions proceeded viaNMRP, and the monomers were taken into the alkoxyamine at the cross-linking points. In the case of styrene and PEGSt, the mesh size became larger, whereas DVB made the mesh size smaller. One advantage of this gel is that the mesh size of chemical gels can be easily controlled from one type of gel by means of monomer selection and reaction time even after the preparation of the gels. Furthermore, variations in gel properties by functional monomer insertions were shown by the swelling degree, and the gel structures were shown by SAXS profiles. These reorganizable chemical gels combine the advantages of the high stability of chemical gels with the structural changeability of physical gels, and this concept will be applicable not only to functional gels, but also more complex molecular systems.Acknowledgements
The authors gratefully acknowledge the financial support of a Grant-in-Aid for Scientific Research (20350057) from the Ministry of Education, Culture, Science, Sports and Technology of Japan. Y.A. acknowledges the financial support of a Grant-in-Aid for JSPS Fellows. The synchrotron radiation experiments were performed at a BL40B2 in the SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2009A1011).Notes and references
- T. Tanaka, S. T. Sun, Y. Hirokawa, S. Katayama, J. Kucera, Y. Hirose and T. Amiya, Nature, 1987, 325, 796–798 CrossRef CAS.
- T. Ono, T. Sugimoto, S. Shinkai and K. Sada, Nat. Mater., 2007, 6, 429–433 CrossRef CAS.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- S. Maeda, Y. Hara, R. Yoshida and S. Hashimoto, Angew. Chem., Int. Ed., 2008, 47, 6690–6693 CrossRef CAS.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. I. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- A. W. Jackson and D. A. Fulton, Macromolecules, 2010, 43, 1069–1075 CrossRef CAS.
- M. F. Li, K. Yamato, J. S. Ferguson, K. K. Singarapu, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2008, 130, 491–500 CrossRef CAS.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS.
- G. H. Deng, C. M. Tang, F. Y. Li, H. F. Jiang and Y. M. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- T. Endo, T. Suzuki, F. Sanda and T. Takata, Macromolecules, 1996, 29, 3315–3316 CrossRef CAS.
- J. P. Kennedy and K. F. Castner, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 2055–2070 CrossRef CAS.
- T. Oku, Y. Furusho and T. Takata, Angew. Chem., Int. Ed., 2004, 43, 966–969 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS.
- R. Nicolay, J. Kamada, A. Van Wassen and K. Matyjaszewski, Macromolecules, 2010, 43, 4355–4361 CrossRef CAS.
- Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and A. Takahara, Macromolecules, 2009, 42, 8733–8738 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- C. J. Hawker, G. G. Barclay and J. L. Dao, J. Am. Chem. Soc., 1996, 118, 11467–11471 CrossRef CAS.
- A. Studer and T. Schulte, Chem. Rec., 2005, 5, 27–35 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- Y. Amamoto, Y. Higaki, Y. Matsuda, H. Otsuka and A. Takahara, Chem. Lett., 2007, 36, 774–775 CrossRef CAS.
- Y. Amamoto, Y. Higaki, Y. Matsuda, H. Otsuka and A. Takahara, J. Am. Chem. Soc., 2007, 129, 13298–13304 CrossRef CAS.
- Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and A. Takahara, Macromolecules, 2010, 43, 1785–1791 CrossRef CAS.
- Y. Amamoto, M. Kikuchi, H. Otsuka and A. Takahara, Macromolecules, 2010, 43, 5470–5473 CrossRef CAS.
- Y. Amamoto, T. Maeda, M. Kikuchi, H. Otsuka and A. Takahara, Chem. Commun., 2009, 689–691 RSC.
- Y. Higaki, H. Otsuka, T. Endo and A. Takahara, Macromolecules, 2003, 36, 1494–1499 CrossRef CAS.
- B. Zhao, D. J. Li, F. J. Hua and D. R. Green, Macromolecules, 2005, 38, 9509–9517 CrossRef CAS.
- A. Onuki, J. Phys. II, 1992, 2, 45–61 CrossRef CAS.
- M. Shibayama, Macromol. Chem. Phys., 1998, 199, 1–30 CrossRef CAS.
- H. D. Bale and P. W. Schmidt, Phys. Rev. Lett., 1984, 53, 596–599 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |