Synthesis of amphiphilic poly(cyclooctene)-graft-poly(ethylene glycol) copolymersviaROMP and its surface properties
Hengchong
Shi
a,
Dean
Shi
*b,
Zhanhai
Yao
a,
Shifang
Luan
a,
Jing
Jin
a,
Jie
Zhao
a,
Huawei
Yang
a,
Paola
Stagnaro
c and
Jinghua
Yin
*a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, P. R. China. E-mail: yinjh@ciac.jl.cn
bMinistry-of-Education Key Laboratory for the Green Preparation and Application of Functional Materials, Faculty of Materials Science and Engineering, Hubei University, Wuhan, 430062, P. R. China. E-mail: deanshi2001@yahoo.com
cIstituto per Io Studio delle Macromolecole, Consiglio Nazionale delle Ricerche, Via de Marini 6, Genova, 16149, Italy
First published on 14th December 2010
Abstract
Macromonomer cyclooctene-poly(ethylene glycol) (cyclooctene-PEG) was first synthesized before being copolymerized with cyclooctene by ring opening metathesis polymerization (ROMP) to obtain an amphiphilic graft copolymer (poly(cyclooctene)-g-PEG) with polycyclooctene as the hydrophobic trunk chain and PEG as hydrophilic side chains. The structure of poly(cyclooctene)-g-PEG copolymer was characterized by FTIR and 1H-NMR. The surface properties of poly(cyclooctene)-g-PEG film were evaluated through water contact angle and X-ray photoelectron spectroscopy (XPS). Water contact angle decreased from 87.7° to 65.8° along with increasing the content of PEG. Protein adsorption results showed that poly(cyclooctene)-g-PEG copolymers had significant effect on preventing bovine serum albumin (BSA) from absorbing onto the polymer surface.
Introduction
Poly(ethylene glycol) (PEG) has been paid much attention due to its hydrophilicity, good solubility in water and organic solvents, non-toxicity, absence of antigenicity and immunogenicity. It has been widely applied in biotechnical and biomedical fields.1–11 Poly(L-histidine)–poly(ethylene glycol) diblock copolymers (polyHis–b–PEG) was synthesized and used for the construction of polymeric micelles in human body.7 Michel et al.8 used poly(L-lysine)-g-poly(ethylene glycol) (PLL-g-PEG) copolymers with different architectures to adsorb onto niobium pentoxide-coated silicon wafers and characterized its surface before and after protein adsorption. Pan et al.9 prepared a series of PEG conjugated PAMAM dendrimers by varying the substitution degrees of the dendrimer surface functional group with PEG. The encapsulation efficiency and the in vitro release characteristics of these PEG conjugated PAMAM dendrimers were studied. Lutz et al.10–13 prepared a series of well-defined graft copolymers with hydrophobic trunk chain and PEG side chains. These polymers can be used to prepare a wide variety of modern materials such as biosensors, smart gels for chromatography, artificial tissues, and drug carriers.There are three paths to synthesize amphiphilic graft copolymers. Path I, the “grafting from” approach:14,15 the side chains are grown from the polymeric backbone pending initiating groups by a variety of controlled polymerization methods such as atom transfer radical polymerization (ATRP), nitroxide-mediated polymerization (NMP) or reversible addition fragmentation chain transfer (RAFT). Path II, the “grafting through” approach:16,17 macromonomers are synthesized first and then homopolymerized or copolymerized with other monomers by ATRP, NMP and RAFT. Path III, the “grafting onto” approach:18–20 two kinds of homopolymers are first prepared separately. One has a lot of reactive sites along with the backbone and the other has functional group at the end of molecule chain. Copolymers are formed via coupling reaction between them.
In Path I, the grafting point can be easily controlled and the purification of copolymers from the reaction system is easy. Although the chain length of the side chain is adjustable in Path III, the grafting site and grafting degree cannot be controlled. However, both the chain length and position of the grafted chains can be easily controlled in method Path II. So in this article, Path II was used to prepare the poly(cyclooctene)-g-PEG amphiphilic graft copolymer. Ring opening metathesis polymerization (ROMP) was adopted with a cyclic monomer (cyclooctene) as the initial point. The surface properties were characterized by water contact angle analysis and X-ray photoelectron spectroscopy (XPS). Finally, protein adsorption experiments were performed on the surfaces of the amphiphilic poly(cyclooctene)-g-PEG copolymer and polycyclooctene, respectively. The results showed that poly(cyclooctene)-g-PEG had a significant effect on the reduction of the adsorption of bovine serum albumin (BSA). These amphiphilic polyclooctene-g-PEG copolymers have potential applications in drug delivery, implant, biomedical coating and so on.
Experimental
Chemicals
Cis-cyclooctene was purchased from Acros Organics Co. Ltd. Lithium aluminium hydride (98.3%) and m-chloroperoxybenzoic acid (85.3%) were purchased from Tianjin Hainachuan Science and Technology Company. 1,5-Cyclooctadiene (99%), 1-hexene (97%), ethyl vinyl ether (99%) and Grubbs' generation II catalyst were purchased from Aldrich. Toluene diisocyanate (TDI) was purchased from Shanghai Lingfeng Chemical Regent Co. Ltd. Poly(ethylene glycol) methylether (Mn = 750) was obtained from Aldrich. Tetrahydrofuran (THF) was distilled with sodium and ketyl benzophenone, and dichloromethane was distilled with calcium hydride. The protein, bovine serum albumin (BSA), was obtained from Beijing Dingguo Biotech. Co. Ltd.Synthesis of cyclooctene-PEG macromonomer
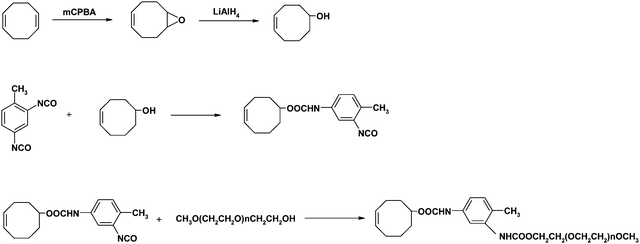
The synthesis procedure for cyclooctene-PEG macromonomer is shown in Scheme 1. 5-Hydroxy-1-cyclooctene was firstly synthesized according to the literature procedure reported earlier.21,22 Then 5-hydroxyl-1-cyclooctene (0.004 mol) was dissolved in 7.5 mL dry toluene and added dropwise to a solution of TDI (0.0042 mol) in 10 mL of toluene at 40 °C under argon atmosphere for 1 h. The reaction was stirred for an additional 12 h to obtain cyclooctene-TDI. Then the temperature was heated to 85 °C. Poly(ethylene glycol) methylether (0.004 mol) in 10 mL toluene was added slowly into the system for 1 h under argon and then reacted until the isocyanate group was completely reacted. Yellow liquid (cyclooctene-PEG macromonomer) was seen when the reaction was completed. The product was purified viacolumn chromatography with CHCl3–CH3OH (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, volume ratio) on silica. The yield of cyclooctene-PEG macromonomer is approximate 60%.
:1, volume ratio) on silica. The yield of cyclooctene-PEG macromonomer is approximate 60%.
 | ||
| Scheme 1 Synthesis route of cyclooctene-PEG macromonomer. | ||
The NMR chemical shifts of cyclooctene-PEG macromonomer were assigned as follows:
1H-NMR
δ (ppm) with CDCl3 as solvent: 7.0 to 7.5 (aromatic-H), 5.5–5.7 (CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) in cyclooctene), 4.6–4.7 (–CH–O in cyclooctene), 3.6 (–O–CH2–CH2– in PEG), 3.38 (CH3–O–PEG).
in cyclooctene), 4.6–4.7 (–CH–O in cyclooctene), 3.6 (–O–CH2–CH2– in PEG), 3.38 (CH3–O–PEG).
The Mn and PDI of cyclooctene-PEG macromonomer are 1085 and 1.02, respectively, according to MALDI-TOF-MS result.
Polymerization procedure
The ROMP route for cyclooctene–PEG macromonomer and cyclooctene is shown in Scheme 2. Cyclooctene–PEG macromonomer (0.05 mmol), cyclooctene (0.95 mmol) and dry dichloromethane (0.8 mL) were added in a dry Schlenk tube under argon. In a separate vial, Grubbs' generation II catalyst (4 μmol) was dissolved with dry dichloromethane (0.2 mL). Both the monomer mixtures and catalyst solution were subjected to two freeze/pump/thaw cycles and then warmed to room temperature. The catalyst solution was rapidly added into the monomer solution and stirred, and then the reaction happened. Upon vitrification, the reaction was terminated using ethyl vinyl ether (1 mL) and about 1 mL CH2Cl2 was added to improve stirring. The product was precipitated into methanol containing 0.01 wt% butylated hydroxyltoluene (BHT) and dried under vacuum after filtration, and 0.12 g (70.9% yield) off-white solid was obtained.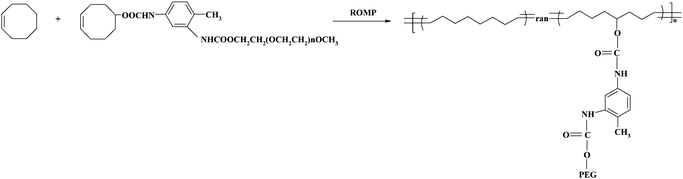 | ||
| Scheme 2 Synthesis route of poly(cyclooctene)-g-PEG. | ||
The NMR chemical shifts of poly(cyclooctene)-g-PEG were assigned as follows:
1H-NMR
δ (ppm) with CDCl3 as solvent: 5.37 (CH2–CH in polymer chain), 4.84 (–CH–O in polymer chain), 4.33 (–COO–CH2–CH2–O), 3.6 (–O–CH2–CH2– in PEG), 3.38 (CH3–O–PEG).
Characterization
1H-NMR spectra were recorded on a Bruker AV 400 MHz spectrometer.Molecular weights and molecular weight distributions were measured on a Waters-2414 gel permeation chromatography (GPC) instrument. The measurements were carried out at 30 °C using CHCl3 as the eluent with a flow rate of 1.0 mL min−1. The system was calibrated with polystyrene standards.
FTIR spectra of both monomers and polymers were recorded using a Bruker Vertex 70 FTIR spectrometer from 4000 to 400 cm−1 at a resolution of 2 cm−1 for 32 scans.
Surfaces for XPS spectroscopy, contact angle measurement, BSA protein adsorption testing were prepared on glass slide by spin-coating 0.5% (w/w) solutions of poly(cyclooctene)-g-PEG graft copolymers and polycyclooctene (virgin) in toluene at 2000 rpm for 30 s using a KW-4A spin coater. Then the surfaces of the samples prepared for measurement were dried in a vacuum oven at reduced pressure at room temperature for at least 12 h to remove solvent completely.
The determination of contact angle was performed on a Drop Shape Analyzer DSA100 (KRÜSS company). A droplet of water (2 μL) was put on the surface of a film and the contact angle was measured. Ten measurements were carried out for a single sample and the values obtained were averaged.
The X-ray photoelectron spectroscopy (XPS) was measured with VG ESCALAB MK at room temperature by using an Mg-Kα X-ray source (hν = 1253.6 eV) at 14 kV and 20 mA. The sample analysis chamber of the XPS instrument was maintained at a pressure of 1 × 10−7 Pa. The takeoff-angle (the angle between the analyzer and the surface normal) was kept at 30° for all samples analyzed. All the C1s peaks were calibrated to the standard binding energy of 284.6 eV for neutral carbon in order to correct the charging energy shifts. A linear-background method removed the XPS background and the peaks analysis carried out by using curve-fitting software.
Protein adsorption test
The BSA was dissolved in the phosphate-buffered saline (PBS, pH ≈ 7.4) at the concentration of 10 mg mL−1. The samples were rinsed initially with PBS and then placed in the BSA solution. The adsorption was allowed to proceed at 25 °C for 24 h. The samples were then removed from the solution, gently washed three times with PBS using a Pasteur pipette, and rinsed once with doubly distilled water to remove the PBS salt. After drying under reduced pressure, the protein-adsorbed surfaces were measured by X-ray photoelectron spectroscopy (XPS). The N1s core-level signal was used as a marker for the analysis of the protein adsorbed on the surface.23Results and discussion
FTIR spectra of cyclooctene–TDI and cyclooctene–PEG macromonomer were shown in Fig. 1. Compared with the curve of cyclooctene–TDI, the strength of the peak at 1701 cm−1 in the curve of cyclooctene–PEG macromonomer which was ascribed to the carbonyl stretching vibration enhanced and the peak at 2273 cm−1 which was NCO stretching vibration disappeared. It showed that the isocyanate group has completely reacted with the hydroxyl group of PEG. A strong peak appeared at 1110 cm−1 for cyclooctene–PEG macromonomer was attributed to the C–O stretch vibration in the PEG group.
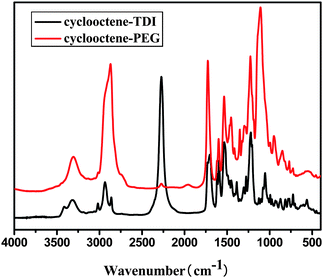 | ||
| Fig. 1 FTIR spectrum of cylooctene–PEG and cyclooctene–TDI. | ||
Fig. 2 is a 1H-NMR spectrum of cyclooctene–PEG macromonomer with CDCl3 as solvent. The peak area ratio between chemical shifts at 5.5–5.7 and 3.38 were attributed to CH2–CH in cyclooctene and CH3– in PEG group and their peak area ratio was 2:3, which was in accordance with the theoretical value.
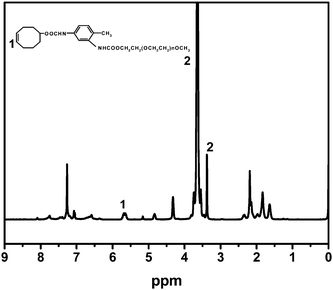 | ||
| Fig. 2 1H-NMR spectrum of cyclooctene–PEG macromonomer. | ||
Graft copolymers 1#–4# were synthesized by ROMP with the appropriate ratio of cyclooctene and cyclooctene–PEG macromonomer in dichloromethane (Table 1), using Grubbs' generation II catalyst. Gel permeation chromatography was used to estimate the molecular weights and molecular weight polydispersity of the graft copolymers by using CHCl3 as eluent. The results are also presented in Table 1. As expected, the polydispersity (Mw/Mn) of all these samples was estimated (by GPC) to be about 2. Although the cyclooctene-PEG macromonomer content in the copolymer calculated by 1H-NMR was a little bit less than its original addition amount, the contents of PEG in these copolymers are still tunable by changing the cyclooctene-PEG macromonomer incorporations.
| Samples | [M]/[Cat] | m cyclooctene-PEG mol (%) | M n × 10−4/g mol−1 | PDI | M cyclooctene –PEG mol (%) |
|---|---|---|---|---|---|
| 1# | 250:1 |
5 | 2.69 | 2.54 | 4.70 |
| 2# | 250:1 |
10 | 1.87 | 2.25 | 9.23 |
| 3# | 250:1 |
15 | 1.21 | 1.76 | 13.87 |
| 4# | 250:1 |
20 | 1.15 | 1.64 | 16.18 |
Fig. 3 displayed the FTIR spectrum of poly(cyclooctene)-g-PEG copolymer. The peak at 1110 cm−1 was attributed to C–O stretching vibration in the poly(ethylene glycol) chains. The peaks at 2925 and 2854 cm−1 were ascribed to the methylene stretch vibration in graft copolymer trunk chain.
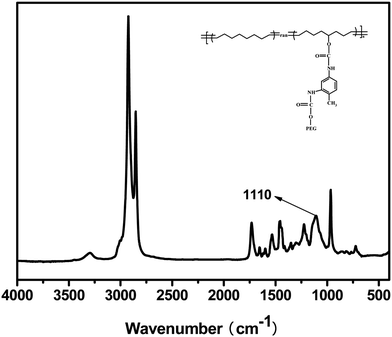 | ||
| Fig. 3 FTIR spectrum of poly(cyclooctene)-g-PEG. | ||
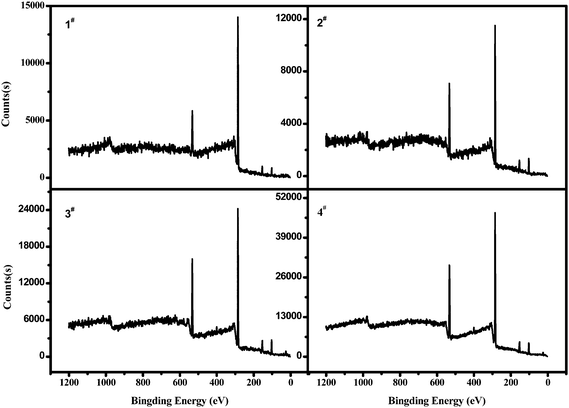
Fig. 4 and Fig. 5 were XPS wide scan spectra of poly(cyclooctene)-g-PEG film surfaces and their corresponding oxygen contents. The PEG graft contents on the film surface, which can be represented by the oxygen contents increased with the increase of the addition amount of cyclooctene-PEG macromonomer. This phenomenon was in accordance with the result that a higher content of PEG grafts in the surface led to a lower contact angle.
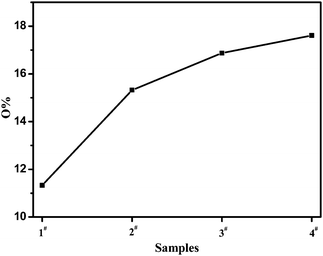 | ||
| Fig. 5 The oxygen content of the poly(cyclooctene)-g-PEG surface. | ||
The water contact angle test is an effective approach for measuring the hydrophility of a polymer surface.15 Lower contact angle values represent higher hydrophilicity. As shown in Fig. 6, the contact angles gradually decreased from 87.7° for polycyclooctene to 65.8° for poly(cyclooctene)-g-PEG copolymer with increasing the PEG contents. The decrease of contact angles should be ascribed to the hydrophilic PEG molecule chains in poly(cyclooctene)-g-PEG copolymers.
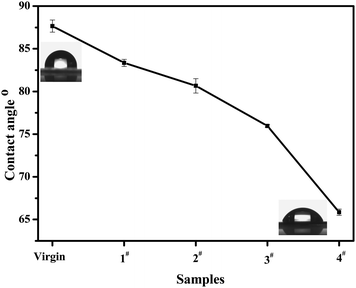 | ||
| Fig. 6 The contact angles of polycyclooctene (virgin) and poly(cyclooctene)-g-PEG (1#–4#). | ||
Bovine serum albumin (BSA) adsorption was one of the methods used to evaluate the biocompatibility of the materials.24 The relative amount of the protein adsorbing on each surface was measured according to XPS measurements. The N1s signal from the peptide bonds was used to mark the relative amount of protein adsorbing on the sample surface. Fig. 7 displayed the XPS N1s core-level spectra of the surfaces of polycyclooctene and poly(cyclooctene)-g-PEG after the protein adsorption in 10 mg mL−1 of BSA buffer solution for 24 h. As shown in Fig. 7, the intensity of the N1s peak component (at the BE of about 400 eV) for the virgin polycyclooctene film surface is much higher than those of poly(cyclooctene)-g-PEG copolymers with different PEG grafting degrees. This strong affinity of the polycyclooctene for BSA is probably due to the hydrophobic interaction of the protein molecules with the highly hydrophobic polycyclooctene surface.25
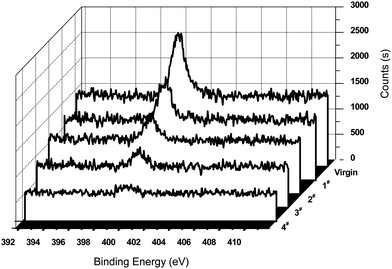 | ||
| Fig. 7 XPS N1s core-level spectra of polycyclooctene (virgin) and poly(cyclooctene)-g-PEG (1#–4#), after exposure to a PBS buffer solution containing 10 mg ml−1 BSA for 24 h. | ||
There are some explanations for reduction of proteins adsorption of poly(cyclooctene)-g-PEG copolymers. The PEG molecules can be highly stretched in water because of its minimum interfacial free energy, hydrophilicity, high surface mobility, steric stabilization effects, unique solution properties and molecular conformation in water.26 These highly stretched PEG molecule chains would prevent protein molecules from approaching the film surface with strong steric exclusion force.27–30 This steric repulsive exclusion force between PEG molecule chains and protein molecules not only comes from the loss of conformation entropy of PEG molecule chains but also relates to the osmotic interaction between them when the protein molecules were approaching to the polycyclooctene surface.29,31,32
4. Conclusions
An amphiphilic graft copolymer (poly(cyclooctene)-g-PEG) was synthesized with polycyclooctene as hydrophobic trunk chain and PEG as hydrophilic side chains through ROMP. The hydrophilic properties of poly(cyclooctene)-g-PEG surface were evaluated through water contact angle and XPS. XPS results also showed that the surface PEG graft contents increased with increasing the addition amount of cyclooctene-PEG macromonomers. Water contact angle decreased from 87.7° for polycyclooctene to 65.8° for poly(cyclooctene)-g-PEG along with the increase of the content of PEG. Protein adsorption results indicated that the PEG grafts in poly(cyclooctene)-g-PEG copolymers had a significant effect on reduction in bovine serum albumin (BSA) adsorption. These amphiphilic polycyclooctene-g-PEG copolymers have potential applications in drug delivery, implants, biomedical coatings and so on.Acknowledgements
The authors are gratefully thankful for the financial support by National Natural Science Foundation of China (Grant no. 50833005, 50920105302), the Key project of Hubei Provincial Department of Education (D20101009) and Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Science 201006. The work has also been carried out in the framework of the Research Cooperation Agreement between the Chinese Academy of Sciences (CAS) and the National Research Council (CNR) of Italy.References
- J. M. Harris, Poly (Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications, Plenum Press, New York, 1992, p. 1 Search PubMed.
- L. D. Unsworth, H. Sheardown and J. L. Brash, Langmuir, 2005, 21, 1036–1041 CrossRef CAS.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, Polym. Chem., 2010, 1, 1205–1214 RSC.
- Y. Dong, P. Gunning, H. Cao, A. Mathew, B. Newland, A. O. Saeed, J. P. Magnusson, C. Alexander, H. Tai, A. Pandit and W. Wang, Polym. Chem., 2010, 1, 827–830 RSC.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Curr. Opin. Colloid Interface Sci., 2001, 6, 3–10 CrossRef CAS.
- H. W. Ma, J. H. Hyun, P. Stiller and A. Chilkoti, Adv. Mater., 2004, 16, 338–341 CrossRef CAS.
- E. S. Lee, H. J. Shin, K. Na and Y. H. Bae, J. Controlled Release, 2003, 90, 363–374 CrossRef CAS.
- R. Michel, S. Pasche, M. Textor and D. G. Castner, Langmuir, 2005, 21, 12327–12332 CrossRef CAS.
- G. Pan, Y. Lemmouchi, E. O. Akala and O. Bakare, J. Bioact. Compat. Polym., 2005, 20, 113–128 CrossRef CAS.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Borner, A. Laschewsky, C. Duschl and J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS.
- J. F. Lutz, H. G. Borner and K. Weichenhan, Macromolecules, 2006, 39, 6376–6383 CrossRef CAS.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- K. L. Beers, S. G. Gaynor, K. Matyjaszewski, S. S. Sheiko and M. Moller, Macromolecules, 1998, 31, 9413–9415 CrossRef CAS.
- M. Zhang, T. Breiner, H. Mori and A. H. E. Muller, Polymer, 2003, 44, 1449–1458 CrossRef CAS.
- J. Klier, A. B. Scranton and N. A. Peppas, Macromolecules, 1990, 23, 4944–4949 CrossRef CAS.
- N. Ayres, Polym. Chem., 2010, 1, 769–777 RSC.
- S. W. Ryu and A. Hirao, Macromolecules, 2000, 33, 4765–4771 CrossRef CAS.
- A. Bousquet, C. Boyer, T. P. Davis and M. H. Stenzel, Polym. Chem., 2010, 1, 1186–1195 RSC.
- H. Shi, D. Shi, L. Yin, J. ShiLuanZhao and J. Yin, React. Funct. Polym., 2010, 70, 449–455 CrossRef CAS.
- M. A. Hillmyer, W. R. Laredo and R. H. Grubbs, Macromolecules, 1995, 28, 6311–6316 CrossRef CAS.
- H. Han, F. Chen, J. Yu, J. Dang, Z. Ma, Y. Zhang and M. Xie, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3986–3993 CrossRef CAS.
- P. Wang, K. L. Tan, E. T. Kang and K. G. Neoh, J. Mater. Chem., 2001, 11, 783–789 RSC.
- Q. Yang, Z. K. Xu, M. X. Hu, J. J. Li and J. Wu, Langmuir, 2005, 21, 10717–10723 CrossRef CAS.
- J. H. Lee, B. J. Jeong and H. B. Lee, J. Biomed. Mater. Res., 1997, 34, 105–114 CrossRef CAS.
- R. Kjellander and E. Florin, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2053–2077 RSC.
- J. M. Harris, Poly (Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications, Plenum Press, New York, 1992, pp. 221–245 Search PubMed.
- J. D. Andrade, V. Hlady and S. I. Jeon, Adv. Chem. Ser., 1996, 248, 51–59 CAS.
- N. V. Churaev, I. P. Sergeeva and V. D. Sonolev, J. Colloid Interface Sci., 1995, 169, 300–305 CrossRef CAS.
- S. R. Sheth and D. Leckband, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8378–8379 CrossRef CAS.
- D. H. Atha and K. C. Ingham, J. Biol. Chem., 1981, 256, 2108–2117.
- J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8399–8404 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |