DOI:
10.1039/C0PY00157K
(Paper)
Polym. Chem., 2011,
2, 685-693
Sequential self-repetitive reaction toward wholly aromatic polyimides with highly stable optical nonlinearity
Received
17th May 2010
, Accepted 30th August 2010
First published on 24th September 2010
Abstract
A sequential self-repetitive reaction (SSRR) based on carbodiimide (CDI) chemistry was utilized for preparing a high-yield wholly aromatic polyimide. The polyimide was synthesized with 4,4′-methylene-diphenylisocyanate (MDI) and a di(acid-ester) compound which was derived from the ring-opening reaction of 3,3′,4,4′-oxydiphthalic dianhydride (ODPA) at room temperature by the addition of equimolar methanol. Poly-CDI was first synthesized from MDI. The di(acid-ester) compound was then reacted with poly-CDI to form poly(N-acylurea). After curing process, N-acylurea moiety was converted to di(ester-amide) structure viaSSRR and further subjected to a ring-closure reaction to form the wholly aromatic polyimide with a Tg of 247 °C. This approach was further taken to prepare thermally stable nonlinear optical (NLO) materials. Similarly a diimide-diacid containing chromophore was reacted with poly-CDI to obtain an intermediate, poly(N-acylurea). The poly(N-acylurea) with the ester side groups would exhibit excellent organosolubility, which enabled the fabrication of high quality optical thin films. After in situ poling and curing processes, N-acylurea moiety was converted to di(ester-amide) structure viaSSRR and further subjected to a ring-closure reaction to form the wholly aromatic NLO polyimide with an electro-optical coefficient, r33 of 25 pm/V (830 nm). Excellent temporal stability at elevated temperatures (200 °C) and a waveguide optical loss of 2.5 dB cm−1 at 1310 nm were also obtained.
Introduction
Polymeric second-order nonlinear optical (NLO) materials have been extensively investigated in the past two decades because of their potential applications in photonic devices, such as optical data transmission, high-speed electro-optic modulators, and optical information processing.1–5 One of the benchmark features for applications is long term NLO stability at elevated temperatures. To achieve this feature, NLO chromophores are usually introduced into high glass transition temperature (Tg) polymers. Consequently, aromatic polyimides have been the materials of choice for realizing excellent NLO stability at elevated temperatures due to their high Tgs. However, commercial applications of polyimides are often limited due to poor solubility and processability, and/or high viscosity even at high processing temperatures. To overcome these shortcomings, research has been focused on the synthesis of organo-soluble polyimides while retaining excellent properties.6–8 Another typical approach is to synthesize poly(amide-imide)9–11 or poly(ester-imide)12–14 by incorporating amide or ester functional groups at regular intervals in the polyimide chains.
Aryl carbodiimide (CDI) can be prepared directly from any aromatic isocyanate. An acid adduct, N-acylurea, readily formed by mixing CDI and carboxylic acids at ambient temperature, can be thermally rearranged to form amide and highly reactive isocyanate (NCO) at temperatures higher than 120 °C in the absence of a catalyst.15 Furthermore, amide-imide is subsequently derived from N-acylureavia a sequential self-repetitive reaction (SSRR) process. The SSRR process involves a series of repetitive reactions including (i) thermal rearrangement of N-acylurea to form amide and isocyanate; (ii) condensation of isocyanate into CDI in the presence of a catalyst; and (iii) an addition reaction between carboxylic acid and CDI to yield a new N-acylurea. In this process, all CDIs, acids and isocyanates would be exhausted at the end of the reaction cycle to eventually form amides or polyamides.
Based on the SSRR model reaction, amide-imide linkages can be obtained by the reaction between aryl carbodiimide and carboxylic acid at high temperatures.15 Therefore, we set forth to synthesize their corresponding polymers by using this SSRR process. In our previous report,16 poly-carbodiimide (poly-CDI) was first prepared from 4,4′-methylene diphenyl diisocyanate (MDI) using 1,3-dimethyl-3-phospholene oxide (DMPO) as catalyst. Poly-CDI then reacted with the carboxylic acid of trimellitic anhydride (TMA) at room temperature to form poly(N-acylurea). Finally, poly(amide-imide) was achieved by the thermolysis of the poly(N-acylurea) and the imidization of anhydrides and isocyanates at 200 °C (Scheme 1(a)).16 Another approach to the synthesis of poly(amide-imide) can also be successfully attempted via the reaction of one equivalent of poly-CDI and two equivalents of diimide-diacid by SSRR (Scheme 1(b)). Because of this successful approach, the synthesis of a wholly aromatic polyimideviaSSRR seems feasible.
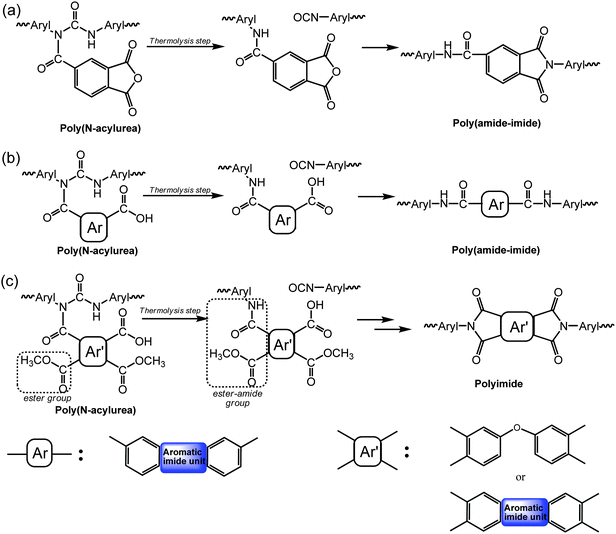 |
| | Scheme 1 The SSRR mechanism for the synthesis of (a) poly(amide-imide) based on TMA, (b) poly(amide-imide) based on diacid-diimide, and (c) polyimide. | |
In this work, wholly aromatic polyimide was synthesized with MDI and a carboxylic acid of the di(acid-ester) compound which was derived from the ring-opening reaction of 3,3′,4,4′-oxydiphthalic dianhydride (ODPA) by the addition of equimolar methanol at room temperature (Scheme 1(c)). The poly(N-acylurea) was prepared from reacting poly-CDI with di(acid-ester) compounds. Through a curing process, the poly(N-acylurea) was transformed into the wholly aromatic polyimidevia two ways: one was by the ring-closing imidization of the ester-amide groups, and the other was by the formation of the ester-amide groups from the reaction between the isocyanate and the carboxylic acid, and subsequently their immediate imidization. We also extend the SSRR process to the preparation a wholly aromatic NLO-active polyimide. This wholly aromatic polyimide is a sensible choice for NLO materials because the reduced number density of N–H bonds in the intrinsic structures plays a key role in lowering the optical losses of materials and its high Tg enhances the stability of dipole orientation at elevated temperatures, when compared to the poly(amide-imide)s mentioned earlier.16,17 It is important to note that the use of N-acylurea as a blocked isocyanate or as an isolable intermediate for polyimide synthesis has not been developed previously.
Experimental
Materials
DMPO (Hochest), TMA (Aldrich), MDI (Acros), PMDA (Lancaster), ODPA (Lancaster) were used as received. DNDA was synthesized according to the literature.16 The solvents were purified by distillation under reduced pressure over calcium hydride.
Preparation of the polyimides
Preparation of the polyimidevia traditional route (PITPIT).
PITPIT
was synthesized according to the literature.18MDI (0.01 mol) was placed in a 100 mL, three-necked, round-bottomed flask equipped with a thermometer, nitrogen gas inlet tube, a reflux condenser, oil bath, and a magnetic stirrer and was dissolved in about 30 mL of dry DMAc; 0.01 mol ODPA was added in small quantities over a period of 10 min at 0 °C with stirring. The solution was stirred at 0 °C for 0.5 h and then slowly heated to 40 °C and held for 2.5 h at this temperature. Subsequently, it was further heated to 90 °C and maintained for 2 h. Carbon dioxide gas evolution was observed at this temperature. The reaction temperature was raised to 130 °C and remained for 20 h. During this period of time, carbon dioxide continued to evolve from the solution, but at a slower rate. A nitrogen atmosphere was maintained in the reaction flask throughout the course of the reaction. The polyimide PITPIT was precipitated by pouring the solution into water. The precipitate was then filtered and washed several times with hot methanol. Then PITPIT was dried in a vacuum oven at 80 °C for 4 h. (yield, 89%) 1H NMR (200 MHz, DMSO-d6) δ = 8.01 (d, ArH, 2H), 7.59–7.33 (m, ArH, 12H), 4.07–3.93 (d, CH2, 2H). FTIR (KBr): 1774 cm−1, 1716 cm−1 (imide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1376 cm−1 and 740 cm−1 (imide ring C–N).
O), 1376 cm−1 and 740 cm−1 (imide ring C–N).
Preparation of the polyimideviaSSRR (PISPIS).
MDI (1 mmol) was dissolved in anhydrous DMAc (10 mL) at room temperature, which was followed by the immediate addition of DMPO (0.01 mL). The solution was stirred at 60 °C for 4 h, and poly-CDI was obtained. Subsequently, ODPE (1 mmol) derived from the ring-opening reaction of ODPA at room temperature by the addition of equimolar methanol, was added to the poly-CDI solution. The solution was kept at 60 °C for another 4 h. After removing DMAc under vacuum, a poly(N-acylurea), PaSPaS was obtained. 1H NMR (200 MHz, DMSO-d6) δ = 8.51 (s, NH, 1H), 7.85 (m, ArH, 2H), 7.30–7.06 (m, ArH, 12H), 3.76 (d, OCH3 and CH2, 8H). To obtain the polyimide PISPIS, the poly(N-acylurea), i.e.PaSPaS was thermally treated at 200 °C for 2 h. SSRR was performed during this period of thermal treatment. 1H NMR (200 MHz, DMSO-d6) δ = 8.01 (d, ArH, 2H), 7.60–7.39 (m, ArH, 12H), 4.07–3.92 (d, CH2, 2H).
Synthesis of NLO polymers
Synthesis of DNDA containing diimide-diacid (11; Scheme 2).
The mixture of DNDA (3.61 g, 0.01 mol) and TMA (3.84 g, 0.02 mol) was dissolved in 10 mL of dry DMAc at room temperature. About 60 mL of toluene was then added, and the mixture was heated with reflux for 4 h until water was distilled off azeotropically. After cooling, the oxblood red precipitate was collected by filtration and washed twice with CH3OH and then dried in vacuum to produce diimide-diacid, 11 (yield, 85%). FTIR (KBr): 3550–3200 cm−1 (carboxyl-OH), 1522 cm−1 and 1362 cm−1 (–NO2), 1782 cm−1, 1726 cm−1 (imide CO), 1366 cm−1 and 726 cm−1 (imide ring C–N). m/z = 710 [M+], C36H19N7O10: Calcd. C, 60.9%; H, 2.7%; N, 13.8%; Found C, 61.1%; H, 3.4%; N, 13.4%.
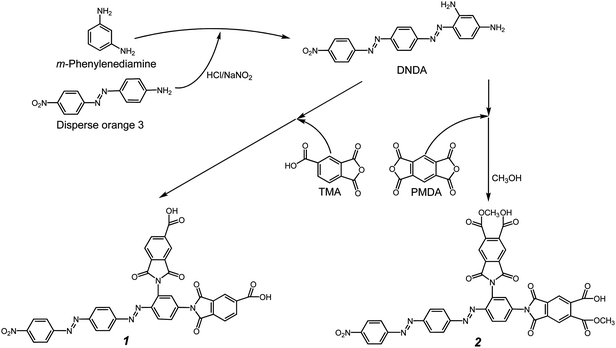 |
| | Scheme 2 Synthesis of DNDA, compounds 11 and 22. | |
Synthesis of DNDA containing diimide-diacid (22; Scheme 2).
The mixture of DNDA (3.61 g, 0.01 mol) and PMDA (8.72 g, 0.04 mol) was dissolved in 15 mL of dry DMAc at room temperature. About 60 mL of toluene was then added, and the mixture was heated with reflux for 4 h until water was distilled off azeotropically. After cooling and adding dry methanol, the oxblood red precipitate was collected by filtration and washed twice with CH3OH–H2O (1/1) and then dried under vacuum to produce diimide-diacid, 22 (yield, 71%). 1H NMR (200 MHz, DMSO-d6) δ = 8.45 (d, ArH, 2H, J = 8.6 Hz), 8.28 (d, ArH, 2H, J = 6.6 Hz), 8.24 (d, ArH, 2H, J = 6.6 Hz), 8.11–8.06 (m, ArH, 5H), 7.96 (d, ArH, 2H, J = 8.6 Hz), 7.95 (s, ArH, 1H), 7.86 (s, ArH, 1H), 3.87 (s, –CH3, 6H). FTIR (KBr): 3550–3200 cm−1 (carboxyl-OH), 1522 cm−1 and 1362 cm−1 (–NO2), 1782 cm−1, 1728 cm−1 (imide CO), 1343 cm−1 and 737 cm−1 (imide ring C–N). m/z = 826 [M+], C40H23N7O14: Calcd. C, 58.2%; H, 2.8%; N, 11.9%; Found C, 58.2%; H, 3.6%; N, 11.6%.
Preparation of the poly(N-acylurea)-11 and the poly(amide-imide)-11 (Pa1Pa1, PI1PI1; Scheme 3).
MDI (1 mmol) was dissolved in anhydrous DMAc (10 mL) at room temperature, which was followed by the immediate addition of DMPO (0.01 mL). The solution was stirred at 60 °C for 4 h, and poly-CDI was obtained. Subsequently, compound 11 (1 mmol) was added to the poly-CDI solution. The solution was kept at 60 °C for another 4 h. After removing DMAc under vacuum, poly(N-acylurea), Pa1Pa1 was obtained. The Mn value and polydispersity of Pa1Pa1 were 11387 and 1.17, respectively. To obtain the poly(amide-imide), the poly(N-acylurea) was thermally treated at 200 °C for 2 h. SSRR was performed during this period of thermal treatment.
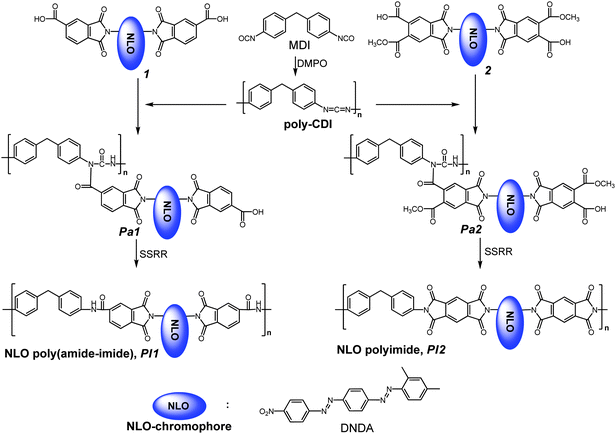 |
| | Scheme 3 Synthesis of Pa1Pa1, Pa2Pa2, PI1PI1 and PI2PI2. | |
Preparation of the poly(N-acylurea)-22 and the polyimide-22 (Pa2Pa2, PI2PI2; Scheme 3).
MDI (1 mmol) was dissolved in anhydrous DMAc (10 mL) at room temperature, which was followed by the immediate addition of DMPO (0.01 mL). The solution was stirred at 60 °C for 4 h, and poly-CDI was obtained. Subsequently, monomer 22 (1 mmol) was added to the poly-CDI solution. The solution was kept at 60 °C for another 4 h. After removing the DMAc under vacuum, a poly(N-acylurea), Pa2Pa2, was obtained. The Mn value and polydispersity of Pa2Pa2 were 34029 and 1.18, respectively. To obtain the polyimide, the poly(N-acylurea) was thermally treated at 200 °C for 2 h. SSRR was performed during this period of thermal treatment.
Preparation of poly(N-acylurea)s and poly(amide-imide-urea)s (PaDN21PaDN21, PIDN21PIDN21, PaDN61PaDN61, PIDN61PIDN61; Scheme 4).
The synthesis of PaDN21PaDN21 and PaDN61PaDN61 samples was performed according to our previous study.16DNDA (1 mmol) was dissolved in anhydrous DMF (5 mL) at room temperature, which was followed by the immediate addition of MDI (6 mmol). The solution was stirred at 60 °C for 2 h. After adding DMPO (0.01mL), poly-CDI was obtained. Subsequently, TMA (5 mmol) was added to the poly-CDI solution. The solution was kept at 60 °C for another 6 h. After removing DMF under vacuum, a polyacylurea, PaDN61PaDN61 was obtained. PaDN21PaDN21 was also prepared in the same manner as PaDN61PaDN61 based on DNDA (1 mmol), MDI (2 mmol) and TMA(1 mmol). The Mn value and polydispersity of PaDN61PaDN61 were 54200 and 3.08, respectively. To obtain the poly(amide-imide-urea), the poly(N-acylurea) was thermally treated at 200 °C for 2 h. SSRR was performed during this period of thermal treatment.
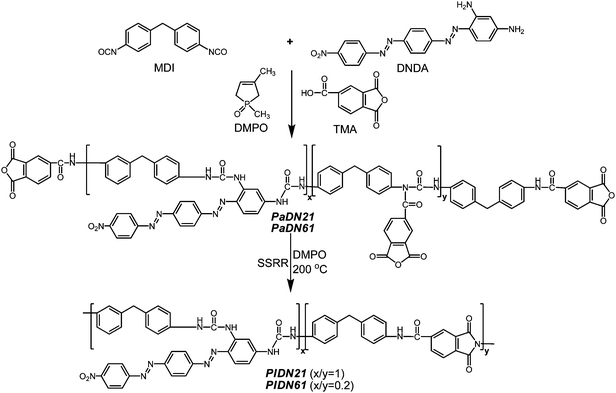 |
| | Scheme 4 Synthesis of PaDN21PaDN21, PaDN61PaDN61, PIDN21PIDN21 and PIDN61PIDN61. | |
Polymer characterization
Infrared spectra were recorded on a Perkin-Elmer Paragon 500 FT-IR spectrophotometer. 1H NMR characterization was carried out on a Varian Gemini200 NMR Spectrometer in DMSO-d6. Elemental analysis (EA) was performed on a F002 Heraeus CHN–O Rapid Elemental Analyzer employing acetanilide as a standard. Fast atom bombardment mass spectrometry (FAB MS) analysis was performed on a JEOL JMS SX/SX 102A mass spectrometer equipped with the standard FAB source, whose upper limit for measuring molecular weight is 2000. A gel permeation chromatograph (Analytical Scientific Instruments Model 500) with a reflection index (RI) detector (Schambeck RI2000) and two columns of Jordi gel DVB mixed bed and 10,000 Å bed in series at 30 °C was used to measure the molecular weight relative to polystyrene standards. The calibration curve was obtained by eight standards in the molecular weight range 3420–2.57 × 106. The carrier solvent was DMF at a flow rate of 1 ml min−1. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were performed on a Seiko SII model SSC/5200 at a heating rate of 10 °C min−1 under nitrogen and air atmosphere, respectively. Thermal degradation temperature (Td) is taken at 5% weight loss. Dielectric analysis (DEA) was taken on an Agilent-4294A impedance analyzer with an applied AC voltage of 1 V. The frequency ranged from 1 kHz to 10 kHz. The polymer films for dielectric analysis were prepared by spin-coating the polymers onto indium tin oxide (ITO) glass substrates. UV-vis spectra of the polymer films were recorded on a Perkin–Elmer Lambda 2S Spectrophotometer.
Thin film preparation
To prepare the second-order NLO polymer films, the poly(N-acylurea)s were respectively dissolved in DMAc. The polymer solution was stirred at room temperature for 3 h, and filtered through a 1 μm filter. Thin films were prepared by spin-coating the filtered polymer solution onto ITO glass substrates. Prior to the poling process, these thin films were dried under vacuum at 60 °C for 24 h. The ellipsometric technique was used here to measure the refractive indices and the thin film thicknesses using a variable angle spectroscopic ellipsometer (VASE), model 2000D from J. A. Woollam Co., Inc. The measurement temperature was maintained at 25 °C. The thicknesses of the polymer films ranged from 0.9 to 1.1 μm, whereas the refraction indices ranged from 1.66 to 1.78.
The poling process, electro-optical (EO) coefficient (r33) and optical loss measurement
The EO coefficients of the poled samples were measured at a wavelength of 830 nm using the simple reflection technique.19 The poling process for the second-order NLO polymer films was carried out using an in situ contact poling technique. The poling voltage was maintained at 70 V and the temperature was kept at 200 °C for 2 h. The formation of the amide-imide and the imide linkages during SSRR process, and the alignment of chromophores proceeded simultaneously during this period. Optical losses of the polymer waveguides for the polymers were measured according to the literature.20 The laser beam (1310 nm) first passed through a linear polarizer and a polarizing beam splitter, thus removing noise caused by polarizing fluctuations in the laser output. Rotation of the linear polarizer allowed the intensity of the beam to be adjusted while rotation of the beam splitter allows for selection of TE polarized light. The un-reflected beam was focused on a prism coupler that is mounted on a rotation stage. The scattered light was imaged with an infrared-sensitive charge-injection-device camera system. A distance scale was provided by imaging a ruler to provide a scale in pixels per centimetre. A statistical linear fit of the data to the logarithm of the scattered light intensity versus the distance propagated down the waveguide yielded a waveguide loss as a slope.
Results and discussion
Two NLO-active chromophores, 11 and 22, both containing two carboxylic acid groups were synthesized by coupling the DNDA with TMA and PMDA, respectively (Scheme 2). Chromophores 11 and 22 were further incorporated into the poly-CDIs, and subsequently formed poly(N-acylurea)s Pa1Pa1 and Pa2Pa2, consisting of the carboxylic acid and acid-ester groups as the side chains, respectively (Scheme 3). The presence of the flexible N-acylurea unit was favorable for the non-centrosymmetic alignment of NLO-active chromophores during the poling/curing process. The intermediates, poly(N-acylurea)s would proceed to undergo SSRR. The thermolysis of the poly(N-acylurea)s yielded the aryl isocyanate and concurrently generated the poly(amide-imide) PI1PI1, and the poly-di(ester-amide) which was further subject to a ring-closure reaction to form the polyimide PI2PI2 as product (Scheme 3). Synthesis and characterization of aromatic polyimidesviaSSRR were discussed as the following.
Monomer synthesis
Chromophore
DNDA was obtained by the azo-coupling reaction of 4-(4′-nitrophenyl-diazenyl)phenyl-1-amine with m-phenylenediamine. DNDA was further incorporated into diimide-diacid monomers via imidization, as shown in Scheme 2.
DNDA containing diimide-diacid monomers 11 and 22 with two preformed imide rings were derived from reacting TMA and PMDA to DNDA, respectively, via a two-stage procedure. This includes ring-opening addition in DMAc to give the intermediate amic acid, and subsequent cyclodehydration to the imidodicarboxylic acid by means of toluene–water azeotropic distillation (Scheme 2). Di(acid-ester) compound 22 was derived from the ring-opening reaction of dianhydrides by the addition of equimolar methanol at room temperature. The yields of diimide-diacid monomers were high, and their structures were confirmed by mass spectrometry analysis, elemental analysis and FT-IR spectra. The FT-IR spectrum of the diimide-diacid monomer 22 showed characteristic imide absorption peaks at 1782, 1728 and 737 cm−1, assigned to symmetrical and asymmetrical CO stretching, and imide ring deformation, respectively, together with characteristic carboxylic acid absorption near 3550–3200 cm−1 (–OH). FT-IR spectrum of compound 11 was also similar to that of compound 22.
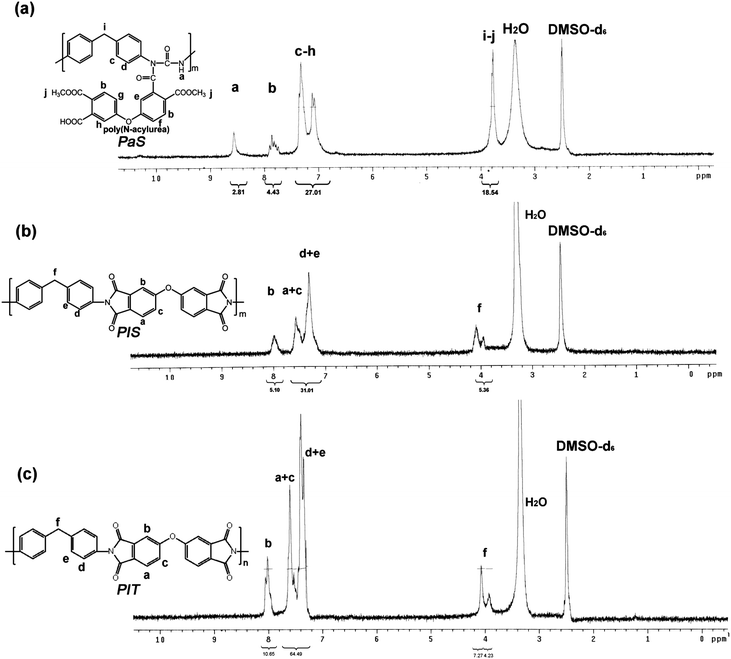
FT-IR spectroscopy was utilized to monitor the SSRR process. As an example, FT-IR spectra of poly-CDI, PaSPaS, PISPIS, and PITPIT are shown in Fig. 1. Poly(N-acylurea) PaSPaS was prepared by the reaction between the poly-CDI and dicarboxylic acid-containing ODPE. In curve (a), the absorption peaks of carbodiimide group were observed at 2112 cm−1 and 2134 cm−1 for the poly-CDI sample. The absorption peaks disappeared as the poly-CDI reacted with ODPE. As shown in curve (b), the absorption peaks of N-acylurea group were observed at 1710 cm−1 for PaSPaS. Subsequently, the thermolysis of the poly(N-acylurea) yielded the aryl isocyanate and concurrently generated the poly-di(ester-amide) which were further subject to a ring-closure reaction to form the polyimide PISPIS as product. For the cured PaSPaS, i.e.PISPIS, the absorption peaks of imide group were observed at 738 cm−1, 1376 cm−1, 1720 cm−1 and 1774 cm−1 as shown in curve (c). The spectrum of PISPIS was similar to that of PITPIT (curve (d)). The FTIR results demonstrate that the polyimide was obtained successfully by the SSRR process. The FT-IR spectra of PI1PI1 and PI2PI2 were also similar to that of PISPIS. Based on the above, it is confirmed that the NLO-active poly(amide-imide) and polyimide were prepared facilely viaSSRR.
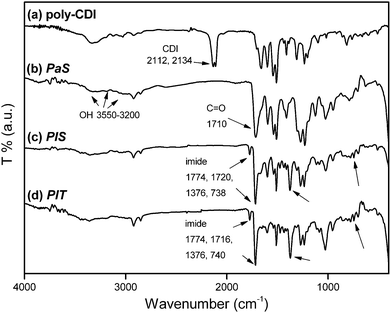 |
| | Fig. 1
Infrared spectra of the (a) poly-CDI, (b) PaSPaS, (c) PISPIS and (d) cured PITPIT. | |
1H NMR spectra were also utilized to monitor the SSRR process during the synthesis of the polyimide. The 1H NMR spectra of PaSPaS, PISPIS and PITPIT in DMSO-d6 are shown in Fig. 2. In the 1H NMR spectrum of PaSPaS, a single peak for the proton of N-acylurea at 8.51 ppm was observed as shown in spectrum (a), indicating that the poly(N-acylurea) was successfully synthesized. For PISPIS, the peak at 8.51 ppm vanished and the peaks for methyl group around 4 ppm emerged (spectrum (b)). The spectrum of PISPIS was similar to that of PITPIT as shown in spectrum (c), indicating the completion of the SSRR process.
The solubility of all poly(N-acylurea) samples PaSPaS, Pa1Pa1 and Pa2Pa2 was excellent in common organic solvents such as THF, DMF, DMAc, and DMSO, especially for PaSPaS and Pa2Pa2 consisting of N-acylurea and carboxyl-ester groups. Because of the excellent organosolubility, these poly(N-acylurea)s could be easily processed into optical quality thin films by spin coating. After the curing process, PISPIS and PITPIT maintained good solubility in DMAc, DMF, DMSO and NMP. Nevertheless, all of the NLO-active polymers exhibited great solvent resistance after SSRR process, especially for the wholly aromatic polyimide PI2PI2. This is possibly because the presence of azobenzene compounds brought about poor solubility in common solvents.21 It is important to note that the polyimide from N-acylurea by SSRR process exhibited excellent processability.
Thermal properties of the polymers
The thermal decomposition behavior was measured under nitrogen using TGA. Td was read at the temperature corresponding to a weight loss of 5%, as summarized in Table 1. The Td values of PISPIS and PITPIT were observed at 544 and 457 °C, respectively. PISPIS possessed a higher Td due to the close stacking of polymer backbone during the heating treatment of the SSRR. As shown in Fig. 3, the Tgs of PISPIS and PITPIT were observed at 247 °C and 217 °C, respectively. This indicates that the presence of the close stacking structure via the process of SSRR enhanced not only the Td of the polymer, but the Tg as well.
Table 1 Thermal properties of the polymers
| Sample |
Chromophore contenta (wt %) |
T
d
b/°C |
T
g
c/°C |
T
g
d/°C |
|
This was further proved by quantitative analysis of the UV-vis spectra of its solution in DMF.
T
d values were defined as weight loss at 5 wt%, which were performed at a heating rate of 10 °C min−1 under nitrogen atmosphere by TGA.
T
g values were performed at a heating rate of 10 °C min−1 under nitrogen atmosphere by DSC.
T
g values were performed at a heating rate of 2 °C min−1 by dielectric analysis.
Not measured.
Not detectable.
|
|
PISPIS
|
0 |
544 |
247 |
—e |
|
PITPIT
|
0 |
457 |
217 |
— |
|
PIDN21PIDN21
|
31 |
266 |
150 |
— |
|
PIDN61PIDN61
|
13 |
277 |
191 |
172 |
|
PI1PI1
|
37 |
290 |
N.D.f |
196 |
|
PI2PI2
|
33 |
377 |
N.D. |
208 |
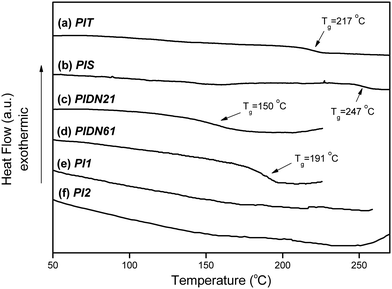 |
| | Fig. 3
DSC thermograms of poly(amide-imide-urea)s, poly(amide-imide) and polyimide: (a) PITPIT, (b) PISPIS, (c) PIDN21PIDN21, (d) PIDN61PIDN61, (e)PI1PI1 and (f) PI2PI2. | |
Thermal properties and NLO chromophore contents are summarized in Table 1. The Td values of the PIDN21PIDN21, PIDN61PIDN61, PI1PI1 and PI2PI2 were observed at 266 °C, 277 °C, 290 °C, and 377 °C, respectively. For the poly(amide-imide) PI1PI1 and the poly(amide-imide-urea)s (PIDN21PIDN21 and PIDN61PIDN61), the Tds were enhanced with increases in the imide content in the polymer backbone. Moreover, the Td of polyimide PI2PI2 was much higher than those of the poly(amide-imide) and poly(amide-imide-urea)s. As shown in Fig. 3, the Tgs of poly(amide-imide-urea) samples, PIDN21PIDN21 and PIDN61PIDN61, were observed at 150 °C and 191 °C, respectively. This indicates that the Tg value was enhanced with increasing imide content in the polymer backbone.16 For PI1PI1 and PI2PI2, their Tgs were not detectable by DSC because the rigid polymer backbone effectively hindered the molecular motion during the glass transition stage. Therefore, the Tgs of PIDN61PIDN61, PI1PI1 and PI2PI2 were measured using a dielectric analyzer at a heating rate of 2 °C min−1. The temperature dependence of dielectric loss tangent for poly(amide-imide-urea)s, poly(amide-imide) and polyimide is shown in Fig. 4. From the dielectric loss tangent thermogram, a relaxation peak corresponding to the glass transition of 172 °C was determined for PIDN61PIDN61.22–24 The relaxation peaks of PI1PI1 and PI2PI2 were determined at 196 °C and 208 °C, respectively. PI2PI2 exhibited lower relaxation intensity than PIDN61PIDN61 and PI1PI1. The lower intensity of dielectric loss tangent peak was attributed to the lower mobility of the rigid polymer chain. In general, the side-chain NLO chromophores would prevent tight packing between the polymer chains, which led to a decrease in relaxation temperature.25 However, PI1PI1 and PI2PI2 possessed higher relaxation temperatures than the PIDN61PIDN61 sample did. This demonstrates that the presence of imide units in the polymer backbone could effectively enhance the relaxation temperature of NLO polymers.
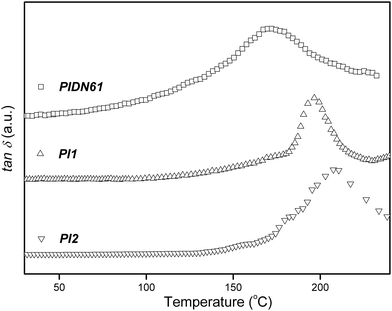 |
| | Fig. 4 Temperature dependence of the dielectric loss tangent for PIDN61PIDN61, PI1PI1 and PI2PI2 films. | |
Optical properties
Optical properties of the NLO polymers are summarized in Table 2. Their refractive indices ranged from 1.66 to 1.78, whereas the EO coefficients (r33) resulting from in situ poling process were in the range between 20 and 25 pm V−1 (measured at 830 nm), dependent on the chromophore density. The EO coefficient of PIDN21PIDN21 was the largest among the poly(amide-imide-urea)-DNDA series samples because of its largest chromophore content. However, the poorest thermal stability (Tg = 150 °C) was observed for the PIDN21PIDN21 sample. Compared with PIDN21PIDN21, the rather thermally stable polymers, PI1PI1 and PI2PI2 exhibited the same EO coefficients due to their similar chromophore contents. This implies that the poling efficiency during the SSRR process was almost the same for PI1PI1 and PI2PI2, despite the fact that the imide segment was more rigid than the amide-imide segment. It is important to note that the alignment of the chromophores was not hindered by the partly formed rigid imide main chain of the PI2PI2 during the SSRR process. Zhou et al.26 and Qiu et al.27 synthesized a series of NLO polyimides from the direct condensation of the di-anhydride and di-amino containing the DNDA chromophore. With around twice as much chromophore content (66 wt.%) as PI2PI2, those polyimides exhibited a relatively smaller EO coefficient (21–23 pm V−1) than PI2PI2 (25 pm V−1 with 33 wt.% of the chromophore content). This indicates that the polyimide formed via the SSRR process exhibited the best poling efficiency of the NLO chromophores. Moreover, the mild SSRR process would prevent the chromophores from decomposing when compared to the conventional harsh imidization process at high temperatures.
Table 2 Optical properties of NLO polymers
| Sample |
Refractive indicesa |
r33b/pm V−1 |
Optical lossc/dB cm−1 |
Thermal dynamic stability/°Cd |
Temporal stability |
| 120 °C 100 h |
200 °C 100 h |
|
Refractive indices were measured at 830 nm wavelength.
EO coefficients were measured at 830 nm wavelength.
Optical losses were measured at 1310 nm wavelength.
Be performed at a heating rate of 2 °C min−1.
Not measured.
|
|
PIDN21PIDN21
|
1.66 |
25 |
3.4 |
—e |
— |
— |
|
PIDN61PIDN61
|
1.75 |
20 |
3.4 |
164 |
15% |
6% |
|
PI1PI1
|
1.77 |
25 |
3.2 |
201 |
81% |
34% |
|
PI2PI2
|
1.78 |
25 |
2.5 |
242 |
98% |
92% |
In general, the optical nonlinearity remains stable at low temperatures, but decays drastically at a specific temperature. This specific temperature is defined as the effective relaxation temperature (Tonset), which allows quick assessment of the temporal and thermal stability of the materials.28 The thermal dynamic stability and temporal stability of EO coefficients were measured only for the samples with better thermal stability (PIDN61PIDN61, PI1PI1 and PI2PI2). Fig. 5 shows the dynamic NLO thermal stability of the poled PIDN61PIDN61, PI1PI1 and PI2PI2 samples. PI1PI1 and PI2PI2 exhibited higher effective relaxation temperatures (201 °C and 242 °C) than PIDN61PIDN61 (Tonset = 164 °C). The Tonset values were enhanced with increasing imide group content. This is due to the fact that the presence of more imide linkages in the polymer chain would further restrict the mobility of the aligned NLO chromophores. The temporal stability of EO coefficients for the poled PIDN61PIDN61, PI1PI1 and PI2PI2 at 120 °C is shown in Fig. 6a. Better temporal stability was obtained for the samples without urea linkages in the polymer chain (PI1PI1 and PI2PI2). In particular, for the PI2PI2 sample, r33 retained 98% of its original value after the thermal treatment for more than 100 h. This is consistent with the dynamic NLO stability study. Apart from the long term NLO stability at approximately 100 °C needed for the commercialization of polymer EO devices, a short excursion to 200 °C is also required.29Fig. 6b shows the temporal stability of EO coefficients for the poled PIDN61PIDN61, PI1PI1 and PI2PI2 at 200 °C. It is important to note that the r33 of the wholly aromatic polyimide (PI2PI2) retained 92% of its original value after thermal treatment at 200 °C for over 100 h. In general, the temporal stability of EO coefficients can be satisfactorily maintained without significant decay only at certain temperatures which are at least 80 °C lower than the Tg of the polymer.30–32 Nevertheless, the wholly aromatic PI2PI2 sample showed commendable temporal stability at temperatures close to its Tg. The presence of wholly aromatic imide linkages in PI2 was indeed capable of hindering molecular motion effectively even at a temperature as high as 200 °C, which is so close to the Tg.
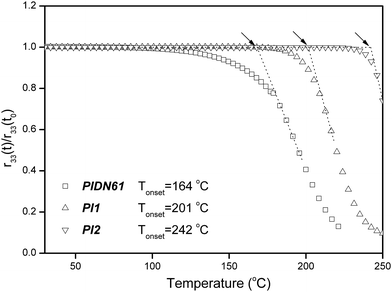 |
| | Fig. 5 Temperature dependence of the dipole re-orientational dynamics of the cured/poled PIDN61PIDN61, PI1PI1 and PI2PI2 samples. | |
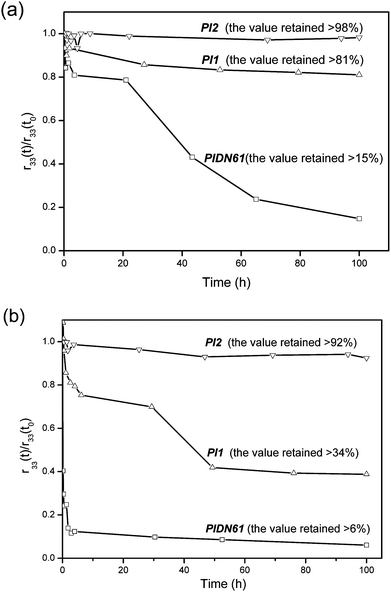 |
| | Fig. 6 Temporal behavior of the EO coefficient for the cured/poled PIDN61PIDN61, PI1PI1 and PI2PI2 samples (a) at 120 °C and (b) at 200 °C. | |
In the investigation of optical losses, the samples were measured at 1310 nm under TE guide modes. As shown in Table 2, the optical losses of these polymer waveguides were in the range 2.5–3.4 dB cm−1. It is important to note that the materials consisting of the urea and amide exhibited larger optical losses than the wholly aromatic polyimide, PI2PI2 did. The reduced optical losses of the thin films might be due to the reduction of the C–H, O–H and N–H number densities.22,33
Conclusion
The synthesis of a wholly aromatic polyimideviaSSRR has been achieved in this work. This novel approach was rather facile when compared with the traditional method using MDI and ODPA. Because of better stacking of the rigid imide linkages between the polymer chains via the SSRR process, the thermal stability was clearly enhanced. A wholly aromatic NLO polyimide also has been developed successfully by SSRR of the poly(N-acylurea). The NLO polyimidevia the SSRR process exhibited a better poling efficiency than the polyimide obtained through the conventional imidization process did. Moreover, this wholly aromatic polyimide showed commendable temporal stability at a temperature close to the Tg. The excellent temporal stability observed was due to the fact that the rigid structure of the wholly aromatic polyimide backbone effectively restricted the molecular motion of the NLO chromophores. Not only was excellent temporal stability at 200 °C obtained, but relatively low optical losses were also achieved for this wholly aromatic polyimide.
Acknowledgements
Financial support from National Science Council of Taiwan and Chung-Shan Institute of Science and Technology is gratefully acknowledged. This work is also supported in part by the Ministry of Education, Taiwan under ATU plan.
Notes and references
- P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2009, 43, 10.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2009, 110, 25.
- J. Luo, X. H. Zhou and A. K. Y. Jen, J. Mater. Chem., 2009, 19, 7410 RSC.
- S. J. Benight, D. H. Bale, B. C. Olbricht and L. R. Dalton, J. Mater. Chem., 2009, 19, 7466 RSC.
- C. C. Chang, C. P. Chen, C. C. Chou, W. J. Kuo and R. J. Jeng, J. Macromol. Sci., Polym. Rev., 2005, 45, 125.
- V. L. Bell, B. L. Stump and H. J. Gager, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 2275 CrossRef CAS.
- J. P. Critchley and M. A. White, J. Polym. Sci., Part A-1, 1972, 10, 1809 CrossRef CAS.
- T. Matsuura, Y. Hasuda, S. Nishi and N. Yamada, Macromolecules, 1991, 24, 5001 CrossRef CAS.
- P. R. Buch and A. V. R. Reddy, Polymer, 2005, 46, 5524 CrossRef CAS.
- G. Maglio, R. Palumbo, R. Schioppa and D. Tesauro, Polymer, 1997, 38, 5849 CrossRef CAS.
- C. Lee, N. P. Iyer, K. Min, H. Pak and H. Han, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 137 CrossRef CAS.
- H. Behniafar and S. Habibian, Polym. Int., 2005, 54, 1134 CrossRef CAS.
- T. Iijima, N. Hayashi, T. Oyama and M. Tomoi, Polym. Int., 2004, 53, 1417 CrossRef CAS.
- N. Song, L. Gao, X. Qiu, W. Qi and M. Ding, Macromol. Chem. Phys., 2000, 201, 1148 CrossRef CAS.
- K. L. Wei, C. H. Wu, W. H. Huang, J. J. Lin and S. A. Dai, Macromolecules, 2006, 39, 12 CrossRef CAS.
- H. L. Chang, H. L. Lin, Y. C. Wang, S. A. Dai, W. C. Su and R. J. Jeng, Polymer, 2007, 48, 2046 CrossRef CAS.
- W. Leng, Y. Zhou, Q. Xu and J. Liu, Macromolecules, 2001, 34, 4774 CrossRef CAS.
- M. Barikani and S. M. Ataei, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2245 CrossRef CAS.
- C. C. Teng and H. T. Man, Appl. Phys. Lett., 1990, 56, 1734 CrossRef CAS.
- R. J. Jeng, G. H. Hsiue, J. I. Chen, S. Marturunkakul, L. Li, X. L. Jiang, R. Moody, C. Masse, J. Kumar and S. K. Tripathy, J. Appl. Polym. Sci., 1995, 55, 209 CrossRef CAS.
- Z. Zhu, Q. Li, Q. Zeng, Z. Li, Z. Li, J. Qin and C. Ye, Dyes Pigm., 2008, 78, 199 CrossRef CAS.
- G. H. Hsiue, R. H. Lee and R. J. Jeng, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2503 CrossRef CAS.
- G. H. Hsiue, R. H. Lee and R. J. Jeng, Polymer, 1999, 40, 6417 CrossRef CAS.
- R. H. Lee, G. H. Hsiue and R. J. Jeng, J. Appl. Polym. Sci., 2001, 79, 1852 CrossRef CAS.
- T. Y. Chao, H. L. Chang, W. C. Su, J. Y. Wu and R. J. Jeng, Dyes Pigm., 2008, 77, 515 CrossRef CAS.
- Y. Zhou, W. Leng, X. Liu, Q. Xu, J. Feng and J. Liu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2478 CrossRef CAS.
- F. Qiu, Y. Cao, H. Xu, Y. Jiang, Y. Zhou and J. Liu, Dyes Pigm., 2007, 75, 454 CrossRef CAS.
- N. Tsutsumi, M. Morishima and W. Sakai, Macromolecules, 1998, 31, 7764 CrossRef CAS.
- S. Kalluri, Y. Shi, W. H. Steier, Z. Yang, C. Xu, B. Wu and L. R. Dalton, Appl. Phys. Lett., 1994, 65, 2651 CrossRef CAS.
- Z. H. Peng and L. P. Yu, Macromolecules, 1994, 27, 2638 CrossRef CAS.
- S. Li, Z. Yang, P. Wang, H. Kang, W. Wu, C. Ye, M. Yang and X. Yang, Macromolecules, 2002, 35, 4314 CrossRef CAS.
- Y. Sui, J. X. Lu, J. Yin, D. Wang, Z. K. Zhu and Z. G. Wang, J. Appl. Polym. Sci., 2002, 85, 944 CrossRef CAS.
- C. P. Chen, G. S. Huang, R. J. Jeng, C. C. Chou, W. C. Su and H. L. Chang, Polym. Adv. Technol., 2004, 15, 587 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.