Synthesis of nitric oxide-releasing polyurethanes with S-nitrosothiol-containing hard and soft segments
Peter N.
Coneski
and
Mark H.
Schoenfisch
*
Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290, USA. E-mail: schoenfisch@unc.edu
First published on 16th February 2011
Abstract
Nitric oxide (NO)-releasing polyurethanes capable of releasing up to 0.20 μmol NO cm−2 were synthesized by incorporating active S-nitrosothiol functionalities into hard and soft segment domains using thiol group protection and post-polymerization modifications, respectively. The nitrosothiol position within the hard and soft segment domains of the polyurethanes impacted both the total NO release and NO release kinetics. The NO storage and release properties were correlated to both chain extender modification and ensuing phase miscibility of the polyurethanes. Thorough material characterization is provided to examine the effects of hard and soft segment modifications on the resultant polyurethane properties.
Introduction
The utility of many biomedical devices is often thwarted by platelet adhesion and thrombus formation that occur soon after contact with blood.1–3For example, biofouling of intravascular implants, such as catheters or stents, may lead to the formation of deep vein thrombosis and increase the risk of heart attack and stroke.2,3 In addition to the potential health hazards caused by surface thrombosis, the costs associated with extended hospital stays, explantations, and re-implantation are rising exponentially.2 Therefore, the design and synthesis of implantable materials that exhibit improved thromboresistivity remains an important goal for researchers developing blood-contacting medical devices.4–6A wide range of synthetic polymers are currently used as medical implants, including polyurethanes, polyesters, and polyacrylates.4,6 In general, more hydrophilic biomaterials possess better blood compatibility due to their low interfacial free energy that reduces plasma protein adsorption relative to hydrophobic implants.7Although polyurethanes are typically composed of both hydrophilic soft segments and hydrophobic hard segments, surface segregation phenomena and surface restructuring upon contact with water provide polyurethanes with the enhanced blood compatibility seen for completely hydrophilic materials.7–10In addition to improved haemocompatibility, the elastomeric and broad mechanical properties provided by the microphase separation of the hard and soft segments make polyurethanes attractive candidates for use in the design of implants.7 Despite these advantages, protein and platelet adhesion to polyurethane surfaces remain problematic in the clinical implementation of devices that use this material.2,3
Nitric oxide (NO) assumes many roles in human physiology including neurotransmission,11,12vasodilation,13,14wound healing,15platelet function,16 and the immune response.17,18 As a result, NO has been investigated as a potential solution to the complications associated with implant biocompatibility.17–20 Unfortunately, the administration of NO is complicated because of its high reactivity and gaseous nature. Consequently, much research has been directed to the development of scaffolds for the storage and controlled release of NO to specific locations.19,21–23 A number of molecular platforms may be used to store NO including organic nitrates, N-diazeniumdiolates, and S-nitrosothiols (RSNOs), the latter being an endogenous NO carrier. RSNO NO donors have received much attention recently due to their low toxicity compared to other NO donor precursors. Furthermore, NO release viaRSNO decomposition is induced by multiple routes including thermal, photolytic and exposure to trace metals.24
Previous reports using NO to improve the thromboresistivity of medical implants made use of NO donors doped into polymer membranes or the direct modification of polymers to release NO.20 While doping of NO scaffolds into polymers allows for tunable NO release, leaching of the NO donor and/or degradation products presents toxicity concerns.25 Others have reported the incorporation of NO-donors (e.g. N-diazeniumdiolates) onto polyurethane scaffolds at pre- and post-polymerization stages.26–28 Unfortunately, the NO release capability is confounded by the presence of protonated surface amines resulting in enhanced protein adhesion.27 Additionally, the surface segregation of the NO donor precursors likely prevents significant NO donor formation (to diazeniumdiolates) due to their location within the hydrophobic hard segments of the polyurethanes.
Herein, we report the synthesized of functional polyurethanes capable of NO storage and controlled release via S-nitrosothiol NO donors. The NO donors are formed after incorporation of the thiols into the polyurethane structure via exposure to acidified nitrite. The influence of RSNO functionalization in both the hard and soft segments of these polyurethanes provides insight into increasing the NO storage capabilities of these materials.
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich (Milwaukee, WI) and used as received unless otherwise noted. Solvents and common laboratory salts were purchased from Fisher Scientific (Philadelphia, PA). Nitric oxide and nitrogen gases were purchased from National Welders Supply (Durham, NC). All water was purified using a Millipore Milli-Q Gradient A-10 purification system (Bedford, MA).Characterization
All 1H and 13C NMR were performed in CDCl3 on a Bruker 400 MHz AVANCE nuclear magnetic resonance spectrometer.Polymer molecular weights were measured using a Waters GPC system with a Wyatt Optilab DSP interferometric refractometer and a Wyatt Dawn EOS as the detector with polystyrene standards. A CAM 200 optical angle goniometer was used to measure static water contact angle. Thermogravimetric analysis was performed on a Perkin-Elmer Pyris 1 TGA under an N2 atmosphere using heating rates of 10 °C min−1. Thermal transitions were measured using a TA Instruments Q200 differential scanning calorimeter with heating rates of 10 °C min−1 and cooling rates of 5 °C min−1. Nitric oxide release was measured using a Sievers 280i nitric oxide analyzer.
General procedure for the S-tritylation of mercaptoacids
Equimolar amounts (30 mmol) of an appropriate mercaptoacid and triphenylmethanol were dissolved in 60 mL of stirring glacial acetic acid, followed by the dropwise addition of 10 mL of boron trifluoride diethyl etherate. After stirring for 4 h at room temperature, the yellow/orange solution and precipitate were poured into a mixture of 150 mL of water and 80 mL of saturated sodium acetate. The aqueous solution was then extracted with diethyl ether. The organic phase was concentrated under reduced pressure and dried in vacuo overnight (Scheme 1). Compound 1a (94% yield). 1H NMR (CDCl3, δ): 3.04 (s, CH2COOH), 7.23 (t, aromatic), 7.31 (t, aromatic), 7.43 (t, aromatic). 13C NMR (CDCl3, δ): 34.58 (CH2COOH), 67.27 (SC(Ph)3), 127.27 (C4), 128.20 (C3), 129.53 (C2), 143.94 (C1), 175.23 (COOH). Anal. calcd for C21H18O2S: C, 75.4; H, 5.4; S, 9.6. Found: C, 75.2; H, 5.5; S, 7.4%. Compound 2a (81% yield). 1H NMR (CDCl3, δ): 1.24 (t, CH3), 3.05 (m, CH2), 3.52 (m, CH3CH), 7.20 (t, aromatic), 7.28 (t, aromatic), 7.43 (t, aromatic). 13C NMR (CDCl3, δ): 15.21 (CH3), 19.71 (CH), 44.15 (CH2), 68.18 (SC(Ph)3), 126.94 (C4), 128.10 (C3), 129.46 (C2), 144.25 (C1), 173.39 (COOH), 173.77 (CONH). Anal. calcd for C24H23NO3S: C, 71.1; H, 5.7; N, 3.5; S, 7.9. Found: C, 67.8; H, 6.0; N, 3.2; S, 6.7%. Compound 3a (51% yield). 1H NMR (CDCl3, δ): 1.20–1.45 (CH2), 1.63 (m, SCH2CH2), 2.17 (t, CH2COOH), 2.38 (t, SCH2), 7.16 (t, aromatic), 7.31 (t, aromatic), 7.44 (t, aromatic). 13C NMR (CDCl3, δ): 24.71 (CH2CH2COOH), 28.66–29.44 (CH2), 32.08 (SCH2), 34.13 (CH2COOH), 66.46 (SC(Ph)3), 126.33 (C4), 127.83 (C3), 129.68 (C2), 145.18 (C1), 180.29 (COOH). Anal. calcd for C30H36O2S: C, 78.2; H, 7.9; S, 7.0. Found: C, 75.4; H, 7.6; S, 6.3%. Compound 4a (98% yield). 1H NMR ((CD3)2SO, δ): 1.95 (t, CH2COOH), 2.07 (SCH2), 7.04 (t, aromatic), 7.12 (t, aromatic), 7.13 (t, aromatic). 13C NMR ((CD3)2SO, δ): 26.97 (SCH2), 33.20 (CH2COOH), 66.49 (SC(Ph)3), 126.94 (C4), 128.23 (C3), 129.37 (C2), 144.62 (C1), 172.93 (COOH). Anal. calcd for C22H20O2S: C, 75.8; H, 5.8; S, 9.2. Found: C, 75.1; H, 5.8; S, 8.3%.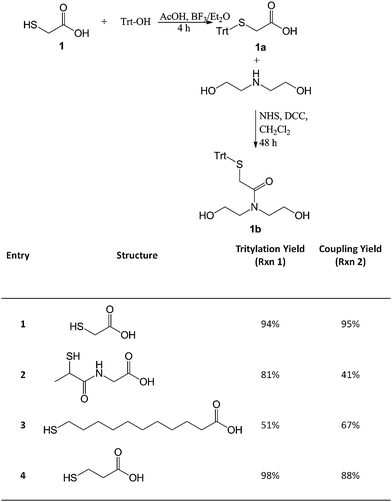 | ||
| Scheme 1 Protected mercaptoacid (1a–4a) and chain extender (1b–4b) synthesis. | ||
General procedure for the coupling of S-trityl mercaptoacids and diethanolamine
A slight excess of N-hydroxysuccinimide (NHS, 22 mmol) was added to a round bottom flask containing the appropriate protected mercaptoacid (20 mmol, 1a–4a) in 80 mL of stirring methylene chloride. After 5 min, the flask was chilled to 0 °C and dicyclohexylcarbodiimide (DCC, 22 mmol) was then added to the solution. The solution was stirred for 10 min on ice before the temperature was allowed to rise to room temperature. The solution was stirred for an additional 24 h under nitrogen. The formed precipitate was removed by vacuum filtration and the filtrate returned to a round bottom flask. Triethylamine (25 mmol) was added to the solution in slight excess followed by the addition of diethanolamine (22 mmol) in methylene chloride (10 mL). After stirring for 24 h under nitrogen, the solution was washed twice with 60 mL of saturated sodium carbonate, and once with 60 mL of saturated sodium bisulfate. The organic phase was then dried over magnesium sulfate, and concentrated under reduced pressure. Compound 1b (95% yield). 1H NMR (CDCl3, δ): 3.04 (m, CH2N), 3.45 (m, CH2OH), 3.77 (t, SCH2), 7.24 (t, aromatic), 7.31 (t, aromatic), 7.47 (t, aromatic). 13C NMR (CDCl3, δ): 33.78 (CH2S), 51.03 (CH2N), 60.84 (CH2COOH), 67.08 (SC(Ph)3), 126.91 (C4), 128.01 (C3), 129.50 (C2), 144.07 (C1), 170.57 (CON). Anal. calcd for C25H27NO3S: C, 71.2; H, 6.5; N, 3.3; S, 7.6. Found: C, 71.6; H, 7.1; N, 4.6; S, 6.2%. Compound 2b (41% yield). 1H NMR (CDCl3, δ): 1.45 (d, CH3), 3.04 (m, CH3CH), 3.36 (t, CH2N), 3.52 (t, CH2OH), 3.80 (CH2CON), 7.20 (t, aromatic), 7.28 (t, aromatic), 7.45 (t, aromatic). 13C NMR (CDCl3, δ): 19.80 (CH3), 33.82 (SCH), 41.89 (NHCH2CON), 50.63 (NCH2CH2OH), 60.13 (NCH2CH2OH), 68.15 (SC(Ph)3), 126.84 (C4), 128.03 (C3), 129.51 (C2), 144.23 (C1), 169.25 (SCH(CH3)CONH), 172.80 (CONCH2CH2OH). Anal. calcd for C28H32N2O4S: C, 68.3; H, 6.6; N, 5.7; S, 6.5. Found: C, 67.4; H, 6.8; N, 5.8; S, 5.9%. Compound 3b (67% yield). 1H NMR (CDCl3, δ): 1.15–1.40 (CH2), 2.15 (m, CH2CON), 2.39 (t, SCH2), 3.52 (t, CH2N), 3.78 (t, CH2OH), 7.22 (t, aromatic), 7.29 (t, aromatic), 7.42 (t, aromatic). 13C NMR (CDCl3, δ): 25.59 (CH2CH2COOH), 28.57–29.23 (CH2), 32.05 (SCH2), 33.92 (CH2COOH), 50.60 (NHCH2CH2OH), 60.85 (NHCH2CH2OH), 66.41 (SC(Ph)3), 126.50 (C4), 127.78 (C3), 129.63 (C2), 145.07 (C1), 175.46 (CON). Anal. calcd for C34H45NO3S: C, 74.6; H, 8.3; N, 2.6; S, 5.9. Found: C, 70.0; H, 8.1; N, 3.3; S, 5.4%. Compound 4b (88% yield). 1H NMR (CDCl3, δ): 2.29 (t, CH2CON), 2.57 (t, SCH2), 3.29 (t, CH2N), 3.65 (t, CH2OH), 7.23 (t, aromatic), 7.30 (t, aromatic), 7.44 (t, aromatic). 13C NMR (CDCl3, δ): 27.42 (CH2S), 33.80 (CH2CON), 50.70 (CH2N), 60.81 (CH2OH), 66.90 (SC(Ph)3), 126.70 (C4) 128.02 (C3), 129.60 (C2), 144.83 (C1), 173.34 (CON). Anal. calcd for C26H29NO3S: C, 71.7; H, 6.7; N, 3.2; S, 7.4. Found: C, 70.9; H, 6.6; N, 3.2; S, 6.8%.Polyurethane synthesis
A round bottom flask was charged with terathane (1 mol eq.), 4,4′-methylenebis(cyclohexyl isocyanate) (4 mol eq.) and dibutyltin dilaurate (0.047 mol eq.) in N,N-dimethylformamide (DMF, 15 mL). The reaction mixture was heated at 75 °C for 90 min followed by the addition of a chain extender (1b–4b, 3 mol eq.) in additional DMF. After a combined reaction time of 24 h at 75 °C, the polyurethane was precipitated in 450 mL water at 4 °C. The solid polymer was isolated by vacuum filtration and then dried in vacuo overnight to yield the protected polyurethane (Scheme 2) (PPU1–PPU4).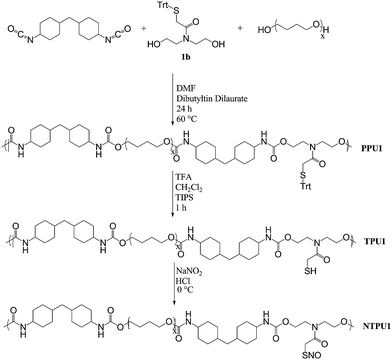 | ||
| Scheme 2 Representative polymerization, deprotection, and nitrosation of TPUpolymers. | ||
Deprotection of protected thiol polyurethanes
After dissolving the protected polyurethane (1.0 g) in CH2Cl2 (30 mL), the solution was chilled on ice. A 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 solution of trifluoroacetic acid:methylene chloride:triisopropysilane (7 mL) was then added dropwise. The reaction mixture was stirred for 1 h on ice, concentrated under reduced pressure, and dried in vacuo to yield the deprotected polyurethane (TPU1–TPU4). To minimize disulfide formation, the solid polymer was stored in an inert atmosphere at −20 °C.
:2:1 solution of trifluoroacetic acid:methylene chloride:triisopropysilane (7 mL) was then added dropwise. The reaction mixture was stirred for 1 h on ice, concentrated under reduced pressure, and dried in vacuo to yield the deprotected polyurethane (TPU1–TPU4). To minimize disulfide formation, the solid polymer was stored in an inert atmosphere at −20 °C.
Polymerization of epichlorohydrin
Epichlorohydrin (420 mmol, 38.81 g) was added dropwise to a solution of ethylene glycol (20 mmol, 1.23 g) and boron trifluoride diethyl etherate (400 μmol, 50 μL) in methylene chloride (10 mL) on ice. After addition of the epichlorohydrin, the solution was allowed to slowly rise to room temperature and react for 15 h. The reaction was quenched by the addition of methanol (5 mL). Subsequent evaporation of the solvent and unreacted starting materials yielded polyepichlorohydrin (PECH) as a light yellow viscous oil (Scheme 3).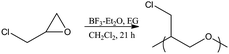 | ||
| Scheme 3 Epichlorohydrin polymerization. | ||
Polymerization of polyepichlorohydrin-containing polyurethane (PU–PECH)
Dibutyltin dilaurate (0.047 mol eq.) in DMF (5 mL) was added to a round bottom flask containing polyepichlorohydrin (1 mol eq.), 4,4′-methylenebis(cyclohexyl isocyanate) (4 mol eq.), and DMF at 70 °C. The solution was stirred for 90 min before 1,4-butanediol (3 mol eq.) was added in additional DMF. After allowing the reaction to proceed for an additional 22.5 h, the polymer was precipitated by pouring the solution into 450 mL of chilled water. The product was then isolated by vacuum filtration (Scheme 4).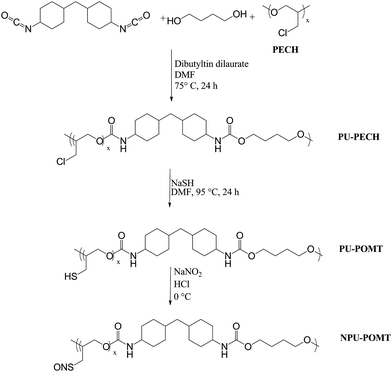 | ||
| Scheme 4 Polymerization, substitution, and nitrosation of PU–PECH. | ||
Thiolation of polyepichlorohydrin-containing polyurethane
Sodium hydrosulfide (4.1 g) was slowly added to a solution of PU–PECH (4.0 g) in DMF (20 mL) at 95 °C. After turning bright blue, the solution was stirred for an additional 24 h, cooled and diluted by the addition of 75 mL CHCl3. The solution was then vacuum filtered to remove insoluble byproducts and concentrated under reduced pressure to yield a polyurethane containing poly(oxiran-2-yl methanethiol) soft segments (PU–POMT).Casting and nitrosation of thiol-containing polyurethane films
Deprotected (TPU1–TPU4) or thiolated (PU–POMT) polyurethane films were solution cast on glass slides from a 40 mg mL−1 solution of polymer in CHCl3, then allowed to dry under ambient conditions for 30 min. Cast films were stored at −20 °C to minimize any further disulfide formation. S-Nitrosothiol functionalized films were prepared by submerging the polymer-coated glass slide in 2 mL of a 50 mg mL−1 solution of NaNO2 in water, and slowly adding 5 mL of 5 M HCl. The films were soaked on ice shielded from light for 2 h, rinsed copiously with a chilled solution of 100 μM diethylenetriamine pentaacetic acid (DTPA) in water, and dried in vacuo. Films were stored at −20 °C prior to experimentation.Nitric oxide release characterization
Nitric oxide release from the polyurethanes was measured using a Sievers model 280i chemiluminescence nitric oxide analyzer (Boulder, CO). The instrument was calibrated using a 26.39 ppm NO gas (balance N2) and air passed through a Sievers NO zero filter. Analysis was performed by placing a polyurethane film in a reaction flask held at 37 °C filled with 30 mL of deoxygenated phosphate buffered saline (PBS) containing 500 μM DTPA and sparged with N2 at a flow rate of approximately 70 mL min−1. Additional N2 flow was supplied via a side-arm to match the collection rate of the instrument at 200 mL min−1. The reaction flask was shielded from light to prevent undesirable photo-triggered release of NO.Results and discussion
Synthesis and characterization of hard segment-modified polyurethanes
The design and synthesis of polyurethanes containing S-nitrosothiol-modified chain extenders or soft segments represent a new class of NO-release scaffold that allows for the evaluation of NO donor spatial effects as a function of thiol position along the backbone. The evaluation of RSNO chain extended polyurethanes first required the synthesis of chain extenders capable of presenting free thiols post-polymerization. To maximize thiol availability following polyurethane synthesis, thiol group protection is essential to prevent thiocarbamate formation upon reaction of thiol-containing chain extenders and isocyanates. As a result, we evaluated the synthesis of four protected thiol-containing chain extenders. The utilization of mercaptoacid starting materials (1–4) provided a facile approach in generating S-trityl mercaptoacids (1a–4a) and subsequent attachment to amine-containing diol species, such as diethanolamine, to generate a group of diverse chain extenders (1b–4b) (Scheme 1).Polyurethane preparation was initiated first by the formation of isocyanate end-capped terathane polyether glycol prepolymers followed by chain extension using the protected thiol chain extenders (1b–4b). NMR analysis of the isolated polymers indicated similar molar ratios of components compared to the reaction mixture. Molecular weight, polydispersity, and thermal characteristics for the resulting protected-thiol polyurethanes (PPU1–PPU4) are shown in Table 1. The molecular weights of the protected thiol polyurethanes ranged from 1.0 × 104 to 1.9 × 104 with polydispersities from 1.4 to 1.9, as expected from step-growth kinetics (PDI ≈ 2.0). The observed glass transition temperature range for the protected polymers (−78 to −64 °C) was similar to values obtained for terathane alone (−77 °C) as predicted by previous studies indicating that polyurethane thermal transitions closely resemble those of their prepolymer derivatives.29 As shown by comparing PPU3 and PPU4 to PPU1, higher glass transition temperatures were observed for polymers containing longer alkanethiol grafts from the chain extenders (−64 and −71 to −78 °C). We attribute this result to the decreased crystallization tendencies of the polyurethanes chains caused by interferences of the chain extender grafts. As the length of these grafts increases, the ability of polymer domains to crystallize is diminished due to spatial interferences of the grafts. As a result, glass transition temperatures increase. Despite these increases, the glass transitions observed indicate that the polyurethanes will maintain their flexibility at physiological temperatures making them suitable candidates for biomaterial applications.
| Sample | M n × 103/g mol−1 | PDI | 5% wt loss/°C | 10% wt loss/°C | T g/°C |
|---|---|---|---|---|---|
| Terathane | 1.2 | 2.1 | 278 | 310 | −77 |
| PPU1 | 12.1 | 1.9 | 238 | 252 | −78 |
| PPU2 | 19.2 | 1.5 | 225 | 241 | −75 |
| PPU3 | 10.0 | 1.7 | 182 | 241 | −64 |
| PPU4 | 12.8 | 1.4 | 216 | 239 | −71 |
| TPU1 | — | — | 142 | 185 | −84 |
| TPU2 | — | — | 112 | 145 | −72 |
| TPU3 | — | — | 88 | 104 | −81 |
| TPU4 | — | — | 131 | 155 | −68 |
| PECH | 1.2 | 1.1 | 279 | 298 | −60 |
| PU – PECH | 4.4 | 1.6 | 171 | 227 | 6 |
| PU – POMT | — | — | 129 | 192 | −9 |
Polyurethanes are desirable biomedical materials due to their unique surface and bulk behavior. For example, the reduced adhesion of blood proteins and platelets is aided by a unique surface reorganization property that occurs in the presence of water.7 In solution, the low surface energy soft segments are oriented at the surface while the hydrophobic hard segments remain in the bulk material to minimize the overall surface free energy at the interface.8 As shown in Fig. 1, PPU1–PPU4 exhibited a surface restructuring phenomenon as evidenced by steadily decreasing water contact angles upon exposure to water droplets. After 20 min in water, all contact angles measured decreased from ∼80° (t = 0) by approximately 1° min−1. Based on this linear trend, the contact angles should all approach that of the polyether glycol alone (44°) after ∼30 min. Due to surface restructuring and a significant concentration of soft segments, the chain extender composition did not have an appreciable effect on the wetting properties of the films.
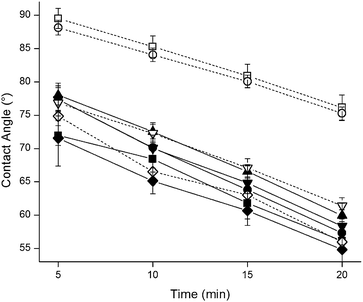 | ||
| Fig. 1 Contact angles of polyurethane samples (■—PPU1, ●—PPU2, ▲—PPU3, ▼—PPU4, ◆—PU–PECH, □—TPU1, ○—TPU2, ▽—TPU4, and ◇—PU–POMT). | ||
Introducing NO-release ability to PPU1–PPU4 required free thiol groups capable of S-nitrosothiol modification. As a result, the polyurethanes were deprotected by exposure to a solution of TFA:CH2Cl2:TIPS for 1 h on ice. Almost immediately, a bright yellow color resulted indicating the formation of the triphenylmethyl cation. Scavenging of this trityl cation by TIPS resulted in a steady decrease in the intensity of the solution's color until it became colorless indicating complete deprotection. Due to the rapid oxidation of thiols to disulfides in the presence of oxygen, special care was taken to minimize oxygen exposure of the isolated thiol-containing polyurethanes (TPU1–TPU4). Still, extremely rapid disulfide formation was observed for TPU3, rendering it insoluble in all organic solvents tested. As the other thiol-containing polyurethanes were not as reactive, the onset of disulfide formation is attributed to the increased chain extender graft mobility (rotational degrees of freedom) about the length of the carbon chain for TPU3 over other TPU compositions. Exposure of the other TPU compositions to ambient oxygen for 1–2 days ultimately resulted in disulfide formation and organic solvent insolubility. The rapid disulfide formation of all TPU conjugates upon exposure to tetrahydrofuran prevented molecular weight determination post-deprotection viaGPC.
Deprotection of the polyurethanes resulted in substantial decreases in degradation temperatures relative to their protected analogs. Thermogravimetric analysis indicated 10% degradation for all TPUs investigated at temperatures at least 67 °C lower than their corresponding protected polyurethane. However, the effect of deprotection on the glass transition temperatures was minimal, with all TPU glass transition temperatures ranging from −68 to −84 °C. These values correspond well with the transition temperature for the polyether glycol soft segment alone making these polymers appropriately flexible materials at physiological temperatures.
Due to decreases in the segmental motion of polymers caused by crosslinking, the influence of spontaneous disulfide formation of TPUs on surface reorganization was a concern. Static water contact angle goniometry was used to characterize the surfaces of thin TPU films exposed to an oxygen atmosphere for at least 5 d. Similar to the PPU family of polyurethanes, all TPU compositions analyzed exhibited static water contact angles that decreased with time indicating that low free energy constituents were able to reorient at the surface of the material. Interestingly, the instantaneous static water contact angles for TPU1 and TPU2 were greater after deprotection than before, indicating an increased presence of hard segment domains at the surface of the material after deprotection. The presence of free thiols in the chain extenders likely acts to promote miscibility of the distinct polymer domains resulting in diminished microphase separation and more uniform hard segment distribution. Differences in the optical clarity of these materials support this observation as the TPU films appeared visibly more opaque than their PPU counterparts that were completely transparent. The static water contact angles vs. time for TPU4 mimicked those of PPU4 more closely than the other polymer analogs did with their deprotected counterparts. The similarity in surface energy is attributed to maintained phase separation as a result of the longer chain extender graft and the phase demixing that remains. Increased optical clarity of TPU4 compared to TPU1 and TPU2 further supports this hypothesis. Overall, the continued presence of domain restructuring at the material:water interface after deprotection for TPU1, TPU2, and TPU4 further ensures biocompatible material characteristics for implant coating materials.
Synthesis and characterization of soft segment-modified polyurethanes
Successful modification of a polyurethane soft segment requires the use of an appropriate polymer with reactive functional groups along the backbone. Isocyanate–alcohol coupling to generate urethane linkages must be retained, necessitating that subsequent functionalization steps not alter or consume the reactive chain termini. The reactivity of functional groups capable of storing NO (e.g., thiols and amines) with isocyanates prevents their incorporation in the backbone prior to polymerization without appropriate protecting groups (similar to PPU1–PPU4) due to the high occurrence of undesirable side reactions and cross-linking. To enable derivatization with NO donor functionalities, polyepichlorohydrin (PECH) was chosen as an appropriate soft segment for further modification. Epichlorohydrin may be polymerized cationically in the presence of low molecular weight diols resulting in the formation of dihydroxytelechelic PECH with molecular weights up to approximately 2.5 × 103 g mol−1.30–33 Despite low molecular weight products, molar masses appropriate for polyurethane soft segments and the telechelic nature of the resulting polymers make this synthetic mechanism ideal. The polymerization of epichlorohydrin using boron trifluoride diethyl etherate in the presence of ethylene glycol led to PECH as a viscous transparent oil. The number average molecular weight of this product was approximately 1.2 × 103 g mol−1 based on gel permeation chromatography with a polydispersity index of 1.1 (Table 1). Nuclear magnetic resonance was used to confirm the presence of alcohol termini (Fig. 2). Subsequent end group analysis indicated molecular weights of approximately 1.6 × 103. The GPC-determined molecular weights likely deviated from those determined using end group analysis due to signal overlap of end group protons by those from the polymer main chain.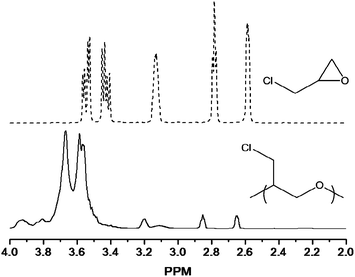 | ||
Fig. 2
1H NMR spectra of epichlorohydrin (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and PECH (—). ) and PECH (—). | ||
Incorporation of the dihydroxytelechelic PECH as a polyurethane soft segment was accomplished by forming the isocyanate end-capped PECHvia reaction in DMF in the presence of dibutyltin dilaurate. The resulting prepolymers were then chain extended using 1,4-butanediol giving rise to PU–PECH with a resulting molecular weight of 4.4 × 103,and PDI of 1.6 (Table 1), and consistent molar ratios of starting materials compared to the reaction solution. The glass transition temperature for PU–PECH (6 °C) was substantially greater than its corresponding soft segment PECH (−60 °C). This deviation is attributed to halogen substitution along the soft segment backbone, a phenomenon previously shown to increase glass transition temperatures.34
The surface properties of PU–PECH follow the trends observed with the chain extender-modified polyurethanes (Fig. 1). Upon exposure to a water droplet for 20 min, the contact angle steadily decreased from 80°, where the hard segment domains were accessible at the surface, to 55°, where the soft segment domains are preferentially oriented at the surface. Similar to the PPUs, the transparency of the films indicated an appreciable degree of microphase separation.
Substitution of the chlorine atoms with thiol functionalities along the polymer soft segment allows for straightforward nitrosation and NO storage. Modification of the soft segment of this polyurethane would also give insight into spatial considerations governing the ability to nitrosate thiols within hard vs. soft segment domains. Thiolation of PU–PECH resulted in a polyurethane, PU–POMT, with similar properties to its parent chain. As expected for a polymer with decreasing chlorine content, the glass transition decreased slightly from 6 °C to −9 °C upon thiolation.34 Similar to the TPUpolymers, the onset of degradation for PU–POMT was also decreased compared to its parent polyurethane.
As a result of the surface segregation phenomenon, incorporation of thiol functionalities along the soft segment domain of polyurethanes should increase the extent of nitrosothiol formation due to increases in the solution accessibility of the thiol containing domain and subsequent nitrosation, which occurs in aqueous solution.8 Additionally, the absence of polar thiol functionalities in the hard segments should prevent mixing of hard and soft segments resulting in elastic materials with a high degree of optical clarity.9 Static water contact angle measurements indicated that thiolation of the polyurethane affected neither the instantaneous surface energy nor the surface reorientation exhibited by most polyurethanes.
Furthermore, the resulting films had a high degree of optical clarity compared to the TPU family of polyurethanes.
Nitrosation and NO release characterization
Enhancing the antifouling behavior of polyurethanes viaNO release represents a significant step toward the development of biocompatible implant coatings. However, the existence of microphase separated domains and surface segregation inherent to polyurethanes makes polymer design a crucial step in the development of a material with appropriate attributes. The design of materials capable of chemically storing NO in both the hard and soft segments of the polyurethane chain should provide insight into spatial considerations necessary for optimal NO donor placement and impact on the material characteristics on the resulting polymer. Nitrosation of the TPUs and PU–POMT was achieved by immersing cast films in a solution of acidified nitrite on ice for 2 h. (Films of TPU3 could not be cast due to disulfide formation; NTPU3 was not investigated.) S-Nitrosothiol formation was confirmed by monitoring the characteristic absorbance using UV-visible spectroscopy24 (Fig. 3).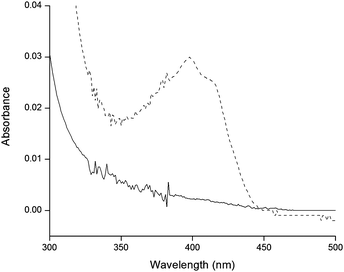 | ||
| Fig. 3
UV/Vis spectra of TPU1 (—) and NTPU1 (). | ||
In accordance with other NO-releasing polymers, the total NO release and maximum instantaneous NO fluxes were found to be highly dependent on the polymer structure (Table 2).26–28After nitrosation, all polymers investigated released NO both in the presence of light (characteristic of S-nitrosothiols) and under physiological conditions (pH 7.4, 37 °C), with NO totals ranging from comparable (see NTPU4, Table 2) to almost an order of magnitude larger (NTPU1 and NPU–POMT) than diazeniumdiolated polyurethanes.27 The levels of total NO release and instantaneous flux followed the same trend for all polymers investigated (NPU–POMT > NTPU1 > NTPU2 > NTPU4). The NO release levels were greatest for NPU–POMT due to the readily accessible thiols along the low surface energy soft segment of the polyurethane, that reoriented outward in solution. The presence of thiols at this interface both facilitated nitrosation by making the functionalities more accessible to solution interactions and allowed for more rapid NO release by minimizing the need for NO diffusion through the polymer matrix.36
| Sample | [NO]T/μmol mg−1 | [NO]T/μmol cm−2 | [NO]max/pmol cm−2s−1 |
|---|---|---|---|
| NTPU1 | 0.11 ± 0.01 | 0.16 ± 0.02 | 532 ± 196 |
| NTPU2 | 0.08 ± 0.03 | 0.11 ± 0.03 | 241 ± 166 |
| NTPU4 | 0.03 ± 0.01 | 0.05 ± 0.01 | 53 ± 4 |
| NPU – POMT | 0.14 ± 0.02 | 0.20 ± 0.03 | 605 ± 253 |
As NO's physiological functions are generally concentration dependent, the duration over which these polymers release NO is an important characteristic.17,35 A nitric oxide flux of >0.4 pmol cm−2s−1 has previously been reported as sufficient for reducing platelet adhesion.35 Although the majority of NO release from the S-nitrosothiol-modified polyurethanes synthesis occurred during the first 10–15 minutes as a bolus, the films continued to release measurable amounts of NO for almost 3 d (NTPU1), with both NTPU1 and NPU–POMT releasing NO above the antithrombotic threshold for >30 h (Fig. 4). Despite releasing NO, NTPU2 and NTPU4 did not maintain antithrombotic levels beyond 1 h.
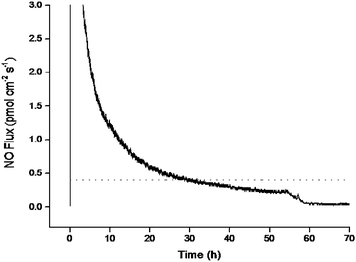 | ||
| Fig. 4 Instantaneous NO flux for NTPU1 (—) in PBS at pH 7.4 and 37 °C compared to antithrombotic threshold values ().35 | ||
As expected, photochemical cleavage of the S–N bond was greatly accelerated compared to the thermal trigger.19,22 In the presence of light, complete NO release from the polyurethanes was observed in less than 15 h compared to over 40 h for materials not exposed to light. Such complete dissociation also makes these materials potentially useful for phototherapy applications.
Despite all NTPU materials possessing thiol-containing hard segments, both the NO release totals and kinetics differed drastically. The increased presence of hard segment domains at the solution interface for TPU1 and TPU2 (due to more efficient domain mixing) allowed for more complete nitrosation due to the solution accessibility of free thiols. The slight NO release disparity between NTPU1 and NTPU2 is likely the result of hard/soft segment miscibility differences between the two polymer systems and the resulting solution accessibility of the hard segments. Indeed, the efficient microphase separation of NTPU4 resulted in a material that released the smallest amount of NO compared to the other polymer systems investigated. The decreased solution accessibility of the hard segments thus limited both thiol to nitrosothiol conversion and NO diffusion through the polymer matrix upon nitrosothiol decomposition.
Conclusion
Surface restructuring and microphase separation are important characteristics of polyurethanes that dictate the behavior of these polymers as implant materials and coatings. It is important to understand that any functionalization of polyurethanes to enhance biocompatibility or performance should not interfere with these properties. S-Nitrosothiol functionalization at hard and soft segment domains of polyurethanes was undertaken to supplement the antifouling behavior of polyurethanes viaNO release and better understand the effects of such modifications on polyurethane properties. Herein, we observed that free thiol incorporation into hard segments alters polyurethane properties based on the resulting phase miscibility of hard and soft segment domains. Additionally, the NO release properties of S-nitrosothiol-modified polyurethanes are related to this domain miscibility with highly miscible domains yielding materials with greater NO storage and release. Soft segment-thiol modification proved to be most promising avenue for NO donor incorporation due to the retention of surface restructuring and microphase separation, and high thiol to nitrosothiol conversion efficiencies related to the solution accessibility of the thiols. Additionally, the NPU–POMT system should provide tunable NO release based on soft segment molecular weight and PECH:chain extender ratios, thereby controlling the total RSNO content. However, the impact of soft segment modifications on the stability and mechanical properties of resulting polyurethanes remains unknown. Although much work has shown the antiplatelet properties of NO-releasing polymers, future studies should evaluate the antifouling properties of these materials.
Acknowledgements
This work was supported by the National Institutes of Health (NIH EB000708). We thank Alexis W. Carpenter for assistance with elemental analysis.References
- J. L. Brash, J. Biomater. Sci., Polym. Ed., 2000, 11, 1135–1146 CrossRef CAS.
- S. R. Hanson, Cardiovasc. Pathol., 1993, 2, 157S–165S.
- B. D. Ratner, Biomaterials, 2007, 28, 5144–5147 CrossRef CAS.
- L. G. Griffith, Acta Mater., 2000, 48, 263–277 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- M. Vert, Prog. Polym. Sci., 2007, 32, 755–761 CrossRef CAS.
- N. Hasirci, in High Performance Biomaterials, ed. M. Szycher, CRC Press, Lancaster, PA, 1991, pp. 71–90 Search PubMed.
- B. F. Pierce, A. H. Brown and V. V. Sheares, Macromolecules, 2008, 41, 3866–3873 CrossRef CAS.
- S. Pongkitwitoon, R. Hernandez, J. Weksler, A. Padsalgikar, T. Choi and J. Runt, Polymer, 2009, 50, 6305–6311 CrossRef CAS.
- R. Navarro, M. Perez, G. Rodriguez and H. Reinecke, Eur. Polym. J., 2007, 43, 4516–4522 CrossRef CAS.
- S. Mocellin, V. Bronte and D. Nitti, Med. Res. Rev., 2007, 27, 317–352 CrossRef CAS.
- R. J. Van't Hof and S. H. Ralston, Immunology, 2001, 103, 255–261 CrossRef CAS.
- R. Schulz, T. Rassaf, P. B. Massion, M. Kelm and J.-L. Balligand, Pharmacol. Ther., 2005, 108, 225–256 CrossRef CAS.
- V. W. T. Liu and P. L. Huang, Cardiovasc. Res., 2008, 77, 19–29 CAS.
- M. B. Witte and A. Barbul, Am. J. Surg., 2002, 183, 406–412 CrossRef CAS.
- M. C. Frost, M. M. Reynolds and M. E. Meyerhoff, Biomaterials, 2005, 26, 1685–1693 CrossRef CAS.
- E. M. Hetrick and M. H. Schoenfisch, Biomaterials, 2007, 28, 1948–1956 CrossRef CAS.
- E. M. Hetrick, H. L. Prichard, B. Klitzman and M. H. Schoenfisch, Biomaterials, 2007, 28, 4571–4580 CrossRef CAS.
- D. A. Riccio, K. P. Dobmeier, E. M. Hetrick, B. J. Privett, H. S. Paul and M. H. Schoenfisch, Biomaterials, 2009, 30, 4494–4502 CrossRef CAS.
- M. M. Reynolds, M. C. Frost and M. E. Meyerhoff, Free Radical Biol. Med., 2004, 37, 926–936 CrossRef CAS.
- J. H. Shin and M. H. Schoenfisch, Chem. Mater., 2008, 20, 239–249 CrossRef CAS.
- N. A. Stasko, T. H. Fischer and M. H. Schoenfisch, Biomacromolecules, 2008, 9, 834–841 CrossRef CAS.
- M. R. Miller and I. L. Megson, Br. J. Pharmacol., 2007, 131, 305–321.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef CAS.
- K. A. Mowery, M. H. Schoenfisch, J. E. Saavedra, L. K. Keefer and M. E. Meyerhoff, Biomaterials, 2000, 21, 9–21 CrossRef CAS.
- H.-W. Jun, L. J. Taite and J. L. West, Biomacromolecules, 2005, 6, 838–844 CrossRef CAS.
- M. M. Reynolds, J. A. Hrabie, B. K. Oh, J. K. Politis, M. L. Citro, L. K. Keefer and M. E. Meyerhoff, Biomacromolecules, 2006, 7, 987–994 CrossRef CAS.
- M. M. Reynolds, J. E. Saavedra, B. M. Showalter, C. A. Valdez, A. P. Shanklin, B. K. Oh, L. K. Keefer and M. E. Meyerhoff, J. Mater. Chem., 2010, 20, 3107–3114 RSC.
- J. Kylma and J. V. Seppala, Macromolecules, 1997, 30, 2876–2882 CrossRef.
- T. Biedron, P. Kubisa and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 2003, 29, 619–628.
- S. Brochu and G. Ampleman, Macromolecules, 1996, 29, 5539–5545 CrossRef CAS.
- S. Carlotti, A. Labbe, V. Rejsek, S. Doutaz, M. Gervais and A. Deffieux, Macromolecules, 2008, 41, 7058–7062 CrossRef CAS.
- A. U. Francis, S. Venkatachalam, M. Kanakavel, P. V. Ravindran and K. N. Ninan, Eur. Polym. J., 2003, 39, 831–841 CrossRef CAS.
- H. J. Oswald and E. T. Kubu, Polym. Eng. Sci., 1963, 3, 168–175 CrossRef CAS.
- M. E. Robbins, E. D. Hopper and M. H. Schoenfisch, Langmuir, 2004, 20, 10296–10302 CrossRef CAS.
- K. A. Mowery and M. E. Meyerhoff, Polymer, 1999, 40, 6203–6207 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |