One-pot enzymatic polycondensation to telechelic methacrylate-functional oligoesters used for film formation
Magnus
Eriksson
ac,
Karl
Hult
c,
Eva
Malmström
b,
Mats
Johansson
b,
Stacy M.
Trey
*ab and
Mats
Martinelle
*c
aSP Trätek, Technical Research Institute of Sweden, Drottning Kristinas väg 67, SE-114 86, Stockholm, Sweden. E-mail: stacy@kth.se; Tel: +46 (0)70 392 6207
bDepartment of Fiber and Polymer Technology, School of Chemical Science and Engineering, Royal Institute of Technology, SE-100 44, Stockholm, Sweden
cDepartment of Biochemistry, School of Biotechnology, Royal Institute of Technology, AlbaNova University Center, SE-106 91, Stockholm, Sweden. E-mail: matsm@biotech.kth.se; Tel: +46 (0)8 55378384
First published on 1st December 2010
Abstract
Based on largely renewable monomers, an enzymatic one-pot polycondensation route towards functional oligomers with targeted molecular weights and end-groups was developed. This one-pot synthesis was performed by combining Candida antarcticalipase B (CALB), 2-hydroxyethyl methacrylate (HEMA), ethylene glycol, and divinyl adipate under reduced pressure (72 mbar) at 60 °C. The polymerization went to completion (>95% conversion for all monomers) within 24 h and the fraction of methacrylate end-groups was >90%. Three targeted dimethacrylate functional oligomers with molecular weights of 920, 1700 and 2500 g mol−1 (degrees of polymerization 4, 8, and 13 respectively) were synthesized. The oligomer products were characterized by NMR, MALDI-TOF MS and SEC. The dimethacrylate functional oligomers were further UV homopolymerized or combined with a tetrathiol crosslinker to demonstrate the potential to produce novel networks with tunable thermal properties dependent on chain length of the telechelic building blocks. This research is the first to demonstrate methacrylate functionalization and condensation polymerization in a one step process, which expands the growing toolbox for polymer/material chemists towards an increased throughput in available macromonomers used in material design.
Introduction
Enzymes have been used as efficient catalysts for the past twenty years to synthesize polymers.1 An important area of polymer synthesis involves controlling the telechelic or end group functionality. By employing the specificity of enzymes, telechelic polyesters have been produced with several different functional end-groups through enzymatic ring-opening polymerization (ROP), see for example.1–6Lipases are frequently used for polyester synthesis through polycondensation with solvent-free systems, even at scaled-up process levels, under mild reaction conditions.1,7,8 The aim of these syntheses has focused more towards polyesters with a high molecular weight than on the introduction of new end-group functional oligomers even though i.e. polyester diols have been reported.7,8 By employing selective enzymatic functionalization strategies in polycondensation routes, an increase in the repertoire of end-functional polycondensation polymers can be envisioned.Our aim is to increase the throughput of telechelic oligomers based on renewable monomers and end-cappers. This calls for simple one-pot processes with high monomer conversion and predictable oligomer products.
The main focus was to develop a method with the potential to generate a large number of polyester macromonomers for polymer network applications. Vinyl esters were chosen in order to achieve a fast route with the added advantage of quantitative conversion of monomers and end-cappers. We hypothesized that by mixing monomers and end-cappers in defined molar ratios, polyesters of desired length and end-group character can be designed. By focusing on producing end-functional oligomers with low molecular weights for subsequent post-polymerization or cross-linking reactions resulting in film formation, efficient chemo-enzymatic routes to new materials based on polycondensation oligomers are accessible. The proposed reaction system has the potential to produce several different telechelic products, made possible by the large number of available building blocks.
The methacrylate group is an important functional group that can effectively be homopolymerized or reacted with thiol groups. Thiol–ene chemistry has been of interest in a broad field of applications including film patterning,9 surface grafting,10 thermoplastics,11 dentistry12 due to industrial advantages including the ability to reach high conversions with little to no photo-initiator in atmospheric conditions. Methacrylated oligomers have been made by enzymatic routes in ROP procedures and by methacrylation of pre-made PEG polymers.3,13
In this research we expand the one-pot enzymatic routes towards telechelic oligomers into polycondensation procedures. This is the first report of the combination of methacrylate functionalizing oligomers in the same step that they are formed by polycondensation. Candida antarcticalipase B (CALB) was used to synthesize telechelic dimethacrylate functional oligomers based on 2-hydroxyethyl methacrylate (HEMA), ethylene glycol and divinyl adipate. The product obtained from the polymerization reaction was a linear, aliphatic polyester containing two methacrylate end-groups. By varying the molar ratio between monomers and end groups it was also possible to obtain oligomers of different desired degree of polymerization (DP). The use of enzymes allows the synthesis of the desired product using one-pot synthesis in a solvent-free system without further purification steps, with the exception of the removal of the enzyme by filtration. The telechelic methacrylate oligomers were subsequently used in a photopolymerization process.
Experimental section
Materials
2-Hydroxyethyl methacrylate (HEMA), 97%, was purchased from Sigma Aldrich (Sweden) and was dried over molecular sieves (3 Å). The immobilized C. antarcticalipase B (Novozyme-435), and deuterated chloroform (CDCl3-d) and dimethyl sulfoxide (DMSO-d6) were purchased from Sigma Aldrich (Sweden). Ethylene glycol (KEBO Lab, Sweden) was dried with sodium sulfate (anhydrous) (from MERCK, Germany) before use. Divinyl adipate (stabilized with MEHQ), 96%, was purchased from TCI Europe (Belgium). Tris(N-nitroso-N-phenylhydroxyl-amine)-aluminium salt (FIRSTCURE® NPAL) was purchased from Albemarle Corporation, USA, and used as a radical inhibitor. The tetra-thiol, pentaerythritol tetra(3-mercaptopropionate) (tSH), was obtained from Bruno Bock Chemische Fabrik GmbH & Co, Germany. The initiator, 2,2-dimethoxy-2-phenylacetophenone (Irgacure 651), was received from Ciba, Sweden, and chloroform was purchased from Fisher Scientific, Sweden. Toluene was purchased from VWR, Sweden.Methods
dMA-PEA-4 . 2-Hydroxyethyl methacrylate (HEMA) (1.15 g, 8.59 mmol), ethylene glycol (800 mg 12.9 mmol) and divinyl adipate (3.55 g, 17.2 mmol) (2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:4 molar ratio) were mixed in a 25 mL round bottom flask (Table 1). N-Nitroso-N-phenylhydroxyl-amine aluminium complex (NPAL, 35 mg) was added as radical inhibitor. The reaction was started by the addition of 100 mg of Novozyme 435 (the trade name for immobilized CALB). The reaction was left under magnetic stirring at 60 °C and allowed to react for 24 h under reduced pressure (72 mbar). The product was dissolved in 10 mL toluene and the immobilized enzyme was removed by a Whatman glass microfiber binder-free filter (grade GF/F). Toluene was evaporated to recover the final oligomer product. NMR and MALDI-TOF-MS analyses were performed on crude samples.
:3:4 molar ratio) were mixed in a 25 mL round bottom flask (Table 1). N-Nitroso-N-phenylhydroxyl-amine aluminium complex (NPAL, 35 mg) was added as radical inhibitor. The reaction was started by the addition of 100 mg of Novozyme 435 (the trade name for immobilized CALB). The reaction was left under magnetic stirring at 60 °C and allowed to react for 24 h under reduced pressure (72 mbar). The product was dissolved in 10 mL toluene and the immobilized enzyme was removed by a Whatman glass microfiber binder-free filter (grade GF/F). Toluene was evaporated to recover the final oligomer product. NMR and MALDI-TOF-MS analyses were performed on crude samples.
| DP a | Ratio HEMA:EG:DAb |
dMA-PEA | dMA-PEA filmc | tSH-PEA filmd |
|---|---|---|---|---|
| a Average degree of polymerization. b HEMA = 2-hydroxyethyl methacrylate, EG = ethylene glycol, DA = divinyl adipate. c Homo-polymerized. d Co-polymerized with pentaerythritol tetra(3-mercaptopropionate) (tSH). | ||||
| 4 | 2:3:4 |
dMA-PEA-4 | dMA-PEA-4 film | tSH-PEA-4 film |
| 8 | 2:7:8 |
dMA-PEA-8 | dMA-PEA-8 film | tSH-PEA-8 film |
| 12 | 2:11:12 |
dMA-PEA-12 | dMA-PEA-12 film | tSH-PEA-12 film |
dMA-PEA-8 . 2-Hydroxyethyl methacrylate (HEMA) (618 mg, 4.60 mmol), ethylene glycol (1.00 g, 16.1 mmol) and divinyl adipate (3.80 g, 18.4 mmol) (2
:7:8 molar ratio) were mixed in a 25 mL round bottom flask and the reaction was performed as described for dMA-PEA-4, with the same amount of Novozyme 435 and reaction time.
dMA-PEA-12 . 2-Hydroxyethyl methacrylate (HEMA) (472 mg, 3.52 mmol), ethylene glycol (1.20 g, 19.3 mmol) and divinyl adipate (4.35 g, 21.1 mmol) (2
:11:12 molar ratio) were mixed in a 25 mL round bottom flask and the reaction was performed as described for dMA-PEA-4, with the same amount of Novozyme 435 and reaction time.
:ene functionality in all films was 1:1. An example formulation is as follows: dMA-PEA-4 (200 mg, equivalent to 0.217 mmol enes), tetra-thiol (tSH) (53 mg, 0.217 mmol thiols), and 2 wt% of Irgacure 651. Films were applied using a 150 µm applicator. Each sample was passed under a UV Fusion Conveyor MC6R equipped with Fusion electrodeless bulbs standard type BF9 lamp five times with a line speed of 1.62 m min−1 to give an overall dose of 1.18 J cm−2. The intensity was determined with a UVICURE® Plus from ET, Sterling, VA. The cured films were transparent with smooth surfaces. The films were kept under vacuum for 24 h at 50 °C after cure.
dMA-PEA . 1H NMR (400 MHz, CDCl3, see Fig. 1 for peak assignment): δ = 1.62–1.73 ppm (4(n + 1)H, m, e), 1.94 ppm (6H, t, b), 2.30–2.40 ppm (4(n + 1)H, t, d), 4.25 ppm (4nH, s, f), 4.35 ppm (8H, s, c), 5.59 and 6.12 ppm (4H, s, a2/a1).
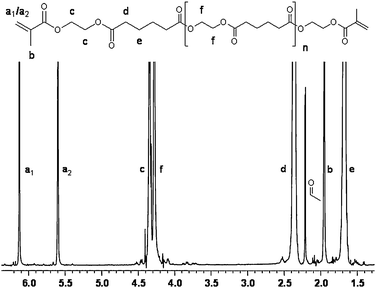 | ||
| Fig. 1 1H NMR spectrum of telechelic dMA-PEA-4. The signal scale has been expanded to show the small peaks in the region 3.5–4.0 ppm. CDCl3 was used as solvent. Acetaldehyde appears at 2.25 ppm. | ||
13C NMR (400 MHz, CDCl3, see Fig. 1 for peak assignment): δ = 18.3 ppm (b), 24.3 ppm (e), 34.0 ppm (d), 62.4 ppm (f), 126.3 ppm (a), 136.2 ppm (δ-carbon of acrylate), 167.1 ppm (carboxyl carbon of methacrylate), 173.3 ppm (carboxyl carbon).
000) from Tosoh Biosep. Further employed were a VE 5200 GPC autosampler, a VE 1121 GPC solvent pump, and a VE 5710 GPC degasser (all from Viscotek Corporation). The SEC apparatus was calibrated with linear polystyrene standards, and toluene was used as an internal standard. The oligomer, 20 mg, was dissolved in 10 mL of warm THF, and filtered with a 0.45 µm Teflon filter before being analyzed.
eqn (1):
 | (1) |
Results and discussion
Enzymatic one-pot bulk synthesis of telechelic dimethacrylate functionalized PEA
This research focuses on the enzymatic synthesis of telechelic methacrylate functional polyesters with targeted DP by polycondensation procedure. A one-pot route was developed by mixing 2-hydroxyethyl methacrylate (HEMA), ethylene glycol and divinyl adipate with C. antarcticalipase B (CALB) at 60 °C under reduced pressure (72 mbar). The conversions of the different reactants were greater than 95% in the crude reaction mixture after 24 h, as determined by 1H NMR. The isolated yield was typically 85–90%, presented in Table 2. From the 1H and 13C NMR it was confirmed that the methacrylate end-groups were intact and bound to the oligomer chain. The two vinyl protons in the methacrylate group (see a1/a2 in Fig. 1) have characteristic signals located at 6.12 and 5.59 ppm, respectively. The methyl group proton signal (see b in Fig. 1) was located at 1.93 ppm. The lack of significant peaks between 3.5 and 4.0 ppm, which correspond to a methylene group adjacent to a terminal hydroxyl group in the involved substrates and oligomer products, showed a very small fraction of free alcohol ends in the oligomers. The vinyl groups in divinyl adipate, with signals at 4.6 and 4.9 ppm in the 1H NMR spectrum, were absent in the products.| Resin | DP a | M n a/g mol−1 | M n (PDI)b/g mol−1 | Yieldc (%) |
|---|---|---|---|---|
| a Determined with 1H NMR (theoretical value within brackets). b Determined by SEC (conventional calibration in THF against polystyrene standard). c Conversion of monomers >95%. | ||||
| dMA-PEA-4 | 4.2 (4) | 920 (900) | 600 (1.9) | 90 |
| dMA-PEA-8 | 8.8 (8) | 1700 (1600) | 1100 (1.9) | 89 |
| dMA-PEA-12 | 13.6 (12) | 2500 (2200) | 2900 (2.5) | 85 |
MALDI-TOF-MS analysis showed a major distribution of peaks, which correspond to the theoretical Mw of the difunctional telechelic oligomer of different degree of polymerization (Fig. 2). The mass difference between the major peaks was 172 Da, corresponding to the repeating unit of ethylene glycol and adipate. The distribution of minor peaks, inset of Fig. 2, correspond to the theoretical weights of oligomer chains containing a monofunctional methacrylate group on one end and, a free acid end group on the other (II), an oligomer chain containing a monofunctional methacrylate group on one end and a sodium carboxylate salt formed from the sodium ion in the MALDI-TOF preparation14 (III), and cyclic chains (IV). The free acid end is a result of the minute amount of water in the system that can hydrolyze the oligomer. In order to demonstrate the simplicity and practicality of the process the reactants were not meticulously dried.
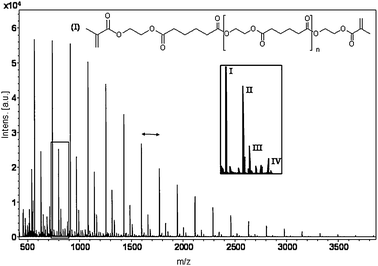 | ||
| Fig. 2 MALDI-TOF-MS spectrum of telechelic dMA-PEA-4. (I) Dimethacrylate product, (II) monomethacrylate functionalized product with an acid end group, (III) monomethacrylate functionalized product with a sodium carboxylate end and (IV) cyclic. Double arrow represents one repeating unit (172 Da). | ||
In order to quantitatively determine the fraction of acid end groups in the oligomer, quantitative 13C NMR analysis was performed. The fraction was calculated by comparing the different signals of the carbonyl carbon in the free acid form (176.2 ppm) and the carbonyl carbon within the bound HEMA end-group (167.1 ppm). For dMA-PEA-4 and -8 the fraction of acid chain ends was determined to be less than 5%, while dMA-PEA-12 had 9% acid ends.
The molar composition is extremely important to reach truly difunctionalized products. In line with step-growth polymerization theory, the variation of end-group structure with a slight error in stoichiometric ratio of reactants becomes relatively greater at higher degrees of polymerization.15 Consequently, the purity of the starting materials has a large impact on the outcome of the reaction.
Molecular weights (Mn) and average DP of the oligomers were determined using 1H NMR (Table 2). Since the oligomers were of low molecular weights the use of NMR to calculate the Mn is a reliable method. Mn and PDI of the different oligomers were also determined by SEC-analysis, results are shown in Table 2. It can be observed from Table 2 that the Mn determined by 1H NMR and by SEC is in good correlation with each other. Small amounts of deviation can be contributed to the polystyrene standards used for calculation, solvent oligomer and end group interactions creating different shapes.15
Further, the proximity of the experimental values in comparison to the targeted values support the conclusion that little to no ethylene glycol reactant was removed under the vacuum conditions of the reaction. Another aspect of the synthesis becomes evident from the data that the PDI becomes broader with increasing molecular weights. This is attributed to the slight error in stoichiometric ratio of reactants, becoming greater at higher degrees of polymerization.16
A reaction system using HEMA, divinyl adipate and 1,4-butanediol, instead of ethylene glycol, was also used. This system revealed the dynamics in enzyme catalyzed oligomer synthesis with acyl transfer reactions present within the produced oligomer (HEMA). The ethylene glycol bound in HEMA migrated throughout the oligomer chain as shown by NMR-analysis. Further variation of the reaction system was tested with the use of 1,4-butanediol dimethacrylate as end-capper. This has shown to be a less favorable substrate in previous work17 and thus, due to slow reaction rates in this study, this monomer was not studied further.
Rational for reaction conditions and reactants
With the aim of demonstrating a clean and scalable route to synthesizing methacrylate functionalized oligomers with a low fraction of cycles, the reaction was performed in bulk under mild conditions. Since employment of CALB in enzyme catalyzed bulk polymerizations has been thoroughly investigated, we decided on this route.2,3,18,19 Novozyme 435 (the trade name for immobilized CALB) has been found to be an efficient catalyst and 3.5% w/w was added to the reaction (with respect to the total amount of monomer employed).1 Vinyl esters were chosen in order to achieve a fast route with the added advantage of quantitative conversion of monomers and end-cappers resulting in clean telechelic oligomers. After testing film properties the synthesis of the selected telechelic oligomers will be developed based on acids or alkyl esters.The reaction was conducted in bulk at 60 °C with magnetic stirring to keep the viscosity of the system low, facilitating homogeneity of the system and good diffusion of reactants. Reduced pressure (72 mbar) was employed during the reaction to remove the water present in trace amounts, since the reactants were not pre-dried. The presence of water will decrease the Mn of the oligomers and result in carboxylic acid end-groups with a reduced fraction of methacrylate functional oligomers. Reduced pressure also removed acetaldehyde, which is the product of the condensation reactions and has a detrimental effect on the enzyme.
HEMA was chosen in order to functionalize the final oligomer enabling it for further reaction for example, thiol–ene systems. Further, HEMA has been previously studied in enzymatic ring opening polymerizations and shown to be a good nucleophile for CALB.20 All components HEMA, ethylene glycol and vinyl adipate have high boiling points allowing for the application of reduced pressure to remove unwanted coproducts, driving the reaction forward.
Film formation, crosslinks and FT-Raman spectroscopy
The oligomers, dMA-PBA-4, -8 and -12, were homo-polymerized or copolymerized by combining them with the tetrathiol as a crosslinker. Six different films were addressed in this study, three homopolymer networks (dMA-PEA films) and three thiol–ene networks (tSH-PEA films). After being exposed to UV light and subsequently cooled, all homopolymer networks and thiol–ene films except those based on dMA-PBA-4 were slightly opaque indicating that crystallization had taken place in the cured films. Homopolymer films, dMA-PBA-8 and dMA-PBA-12, had a smooth non-tacky feel, while dMA-PBA-4 and thiol–ene films had a slight tack to them. No cure was observed during the short flash-off period. Further the occurrence of the Michael addition reaction was insignificant in this case by reason that a weak base is necessary to aid in deprotonation of the thiol groups in order to catalyze this reaction. Further, alkyl methacrylates are reported to be poor Michael acceptors while only strong nucleophiles such as alkyl acrylates or acrylamide groups will readily accept weaker thiol nucleophiles.21FT-Raman was used to monitor the degree of cure, in which characteristic bands of ene and thiol were monitored at 1640 and 2580 cm−1, respectively. Conversion of both the thiol and ene functional groups in the films was determined using eqn (1), in which the spectra were normalized to the unchanging ester group in the system (Table 3). The homopolymer networks (dMA-PEA films) had very high methacrylate conversion except for in the case of dMA-PBA-12 film. This is most likely due to the longer oligomer chains, resulting in a lower functional group concentration to volume, and reduced mobility due to viscosity of the higher Mw chains and their increasing ability to crystallize after being taken out of the oven, in comparison to dMA-PBA-4 and -8 films. The thiol–ene networks may give an indirect indication of how much methacrylate homopolymerization is taking place in this system by observing the amount of thiol remaining in the films.
Overall there is high conversion of the ene with no peak being detected and high conversion of the thiol groups, with small residual amounts due to the favorable methacrylate homopolymerization.22 These results are a contrast to the thiol–allyl ether network research studied by this group in which thiol–allyl ether systems with similar molecules showed very high conversions of both the thiol and the allyl–ether groups, indicating little homopolymerization due to the slower and less favorable reaction of allyl–ether homopolymerization.23
Thermal properties by DSC
The thermal properties of the films were analyzed by DSC, Table 4. The residual crosslinker does not seem to plasticize the film too much as seen by similar Tg values, measured by DSC, for homopolymer networks compared to thiol–ene networks (Table 4). However, the crosslinker does provide star branch points which inhibit crystallization compared to the homopolymerized films.With increasing DP of dMA-PEA polyesters, increasing crystallinity with a Tm ranging from around room temperature for dMA-PEA-4 to 40 °C for dMA-PEA-12 was observed. The crystallinity of dMA-PEA-4-film and tSH-PEA-4-film disappears due to polymerization, resulting in amorphous films. As expected, the crystallinity decreases as the oligomers are homopolymerized, as can be seen by the decrease in the enthalpy of fusion ΔHf, determined from the area of the crystalline Tm exotherm. The larger decrease in crystallinity for dMA-PEA-12-film compared to dMA-PEA-8-film can be attributed to the lower conversion of the thiol monomer, higher PDI, and larger amount of acid end groups (9%) of dMA-PEA-12 (Table 2). There is then a further decrease in the crystallinity of the material once the oligomers are incorporated into thiol–ene networks.
Conclusions
Our priority was to create telechelic difunctionalized polyesters with relatively low molecular weight for use in film formation applications. In this research telechelic polyesters with high fraction (>90%) of methacrylate end-functional groups and targeted molecular weights were synthesized in a simple enzymatic one-pot synthesis. The amount of acid end groups was shown to be low, from 5–9%. High conversion of monomers >95% resulted in clean oligomer products that were used without further purification. The resulting oligomers were then homopolymerized and incorporated into thiol–ene films, in which all films had high conversion of thiol and methacrylate functional groups, resulting in a range of thermal film properties from amorphous (dMA-PEA-4-film and tSH-PEA-4-film) to highly crystalline in the case of homopolymerized dMA-PEA-8-film.This synthesis demonstrates the use of polycondensation to result in methacrylate functional oligomers, being industrially applicable due to the one-pot ease of synthesis and clean up. This type of enzymatically catalyzed polycondensation synthesis towards telechelic methacrylate functionalized polyesters has not been previously reported. The wide variation of building blocks available for polycondensation opens a promising synthetic strategy to new materials with different end-functionalization, backbone structure and oligomer design. Future applications for these products include building blocks for block copolymers, comb polymers and polymer networks, for instance.
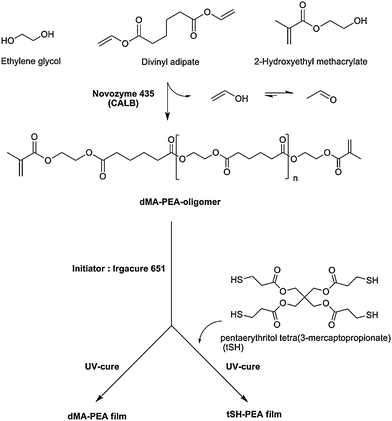 | ||
| Scheme 1 One-pot enzymatic route to dimethacrylate functional poly(ethylene adipate) (dMA-PEA, DP 4, 8, 12), reaction scheme and film synthesis. | ||
Acknowledgements
We thank the Swedish Research Council and BiMaC (Biofibre Materials Centre) for financial support.References
- S. Kobayashi, Macromol. Rapid Commun., 2009, 30, 237–266 CrossRef CAS.
- M. Takwa, N. Simpson, E. Malmström, K. Hult and M. Martinelle, Macromol. Rapid Commun., 2006, 27, 1932–1936 CrossRef CAS.
- M. Takwa, K. Hult and M. Martinelle, Macromolecules, 2008, 41, 5230–5236 CrossRef CAS.
- M. Eriksson, L. Fogelström, K. Hult, E. Malmström, M. Johansson, S. Trey and M. Martinelle, Biomacromolecules, 2009, 10, 3108–3113 CrossRef CAS.
- A. Córdova, T. Iversen and K. Hult, Polymer, 1999, 40, 6709–6721 CrossRef CAS.
- J. W. Peeters, A. R. A. Palmans, E. W. Meijer, C. E. Koning and A. Heise, Macromol. Rapid Commun., 2005, 26, 684–689 CrossRef CAS.
- F. Binns, P. Harffey, S. M. Roberts and A. Taylor, J. Chem. Soc., Perkin Trans. 1, 1999, 2671–2676 RSC.
- F. Binns, S. M. Roberts, A. Taylor and C. F. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1993, 899 RSC.
- V. S. Khire, A. W. Harant, A. W. Watkins, K. S. Anseth and C. N. Bowman, Macromolecules, 2006, 39, 5081 CrossRef CAS.
- A. W. Harant, V. S. Khire, M. S. Thibdaux and C. N. Bowman, Macromolecules, 2006, 39, 1461–1466 CrossRef CAS.
- S. Boileau, B. Mazeaud-Henri and R. Balckborow, Eur. Polym. J., 2003, 39, 1395 CrossRef CAS.
- H. Lu, J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2005, 21, 5301.
- J. E. Puskas, Y. Mustafa and S. Kwang Su Seo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2959–2976 CrossRef CAS.
- H. Yu, E. Lopez, S. W. Young, J. Luo, H. Tian and P. Cao, Anal. Biochem., 2006, 354, 182–191 CrossRef CAS.
- T. Biela, A. Duda and S. Penczek, Macromol. Symp., 2002, 183, 1–10 CrossRef CAS.
- G. Odian, Principles of Polymerization, John Wiley and Sons, Inc., Hoboken, NJ, 4th edn, 2004, p. 76 Search PubMed.
- P.-O. Syrén, E. Lindgren, H.-W. Hoeffken, C. Branneby, S. Maurer, B. Hauer and K. Hult, J. Mol. Catal. B: Enzym., 2009, 65, 3–10.
- I. K. Varma, A. C. Albertsson, R. Rajkhowa and R. K. Srivastava, Prog. Polym. Sci., 2005, 30, 949–981 CrossRef CAS.
- A. Kumar, B. Kalra, A. Dekhterman and R. A. Gross, Macromolecules, 2000, 33, 6303–6309 CrossRef CAS.
- M. Takwa, Y. Xiao, N. Simpson, E. Malmström, K. Hult, C. E. Coning, A. Heise and M. Martinelle, Biomacromolecules, 2008, 9, 704–710 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- Y. L. Tai, Z. Smith, S. K. Reddy, N. B. Cramer and C. N. Bowman, Macromolecules, 2007, 40(5), 1466–1472 CrossRef CAS.
- M. Eriksson, A. Boyer, L. Sinigoi, K. Hult, E. Malmström, M. Johansson, S. Trey and M. Martinelle, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5289–5297 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |