Exploring RAFT polymerization for the synthesis of bipolar diblock copolymers and their supramolecular self-assembly†
Paul E.
Williams
ab,
Adam O.
Moughton‡
ab,
Joseph P.
Patterson
b,
Saghar
Khodabakhsh
c and
Rachel K.
O'Reilly
*b
aMelville Laboratory for Polymer Synthesis, Department of Chemistry, Lensfield Road, Cambridge, CB2 1EW, UK
bUniversity of Warwick, Department of Chemistry, Gibbet Hill Road, Coventry, CV7 4AL, UK. E-mail: R.K.O-Reilly@warwick.ac.uk
cDepartment of Physics, Cavendish Laboratory, J. J. Thomson Avenue, Cambridge, CB2 OHE, UK
First published on 30th November 2010
Abstract
Until recently, the primary controlled radical polymerization (CRP) technique used to synthesize side chain semi-conducting block copolymers from vinyl monomer species has been nitroxide-mediated polymerization (NMP). The potential exploitation of reversible addition fragmentation chain transfer (RAFT) polymerization for the preparation of semi-conducting diblocks has not yet been fully realized. In this work a trithiocarbonate chain transfer agent (CTA) has been shown to polymerize both hole transporting (HT) monomers m-vinyltriphenyl amine and p-vinyltriphenyl amine and also a new fluorinated triphenylamine monomer for the first time, affording both homopolymers and diblock copolymers with good control over molecular weight (Mn) and narrow polydispersities (Mw/Mn). The electronic properties of these blocks and diblocks were explored using UV-vis and cyclic voltammetry analysis. The selective self-assembly of these diblocks into solution nanostructures has been explored and characterized by DLS and TEM analysis.
Introduction
In recent decades conjugated polymers, i.e.polymers with alternating double and single bonds in either the main chain or within the side chain functionalities have been intensely studied due to their novel electronic and optical properties which allow for high semi-conductivity and electroluminescence.1–4 Significant progress has been made in creating organic semi-conducting devices such as light emitting diodes (LEDs), field effect transistors (FETs) and photovoltaics from semi-conducting polymers which have been made by various polymerization techniques.5–11Polymers used to fabricate these devices typically have hole conducting (p-conductor) moieties present. However, if further photovoltaic device optimization is to be realized, both electron (n-conductor) and hole conducting polymers should be incorporated into a single semi-conducting material. For photovoltaic cells, it has been proposed that forming such a p–n heterojunction with two kinds of semi-conductors is necessary to facilitate the dissociation of the photo generated excitons and to prevent recombination in organic thin films. A spatially segregated structure promotes the rapid separation of photo-generated holes and electrons into spatially distinct collection networks and leads to an enhancement of photovoltaic device performance.10,12Supramolecular assemblies of conducting polymers in the 10–100 nm length scale have been suggested to hold great potential for enhanced optoelectronic, photovoltaic activity and may replace inorganic electrical circuitry.13 Such devices have many added advantages over existing non-polymer based technologies such as low cost, solution processability and substrate flexibility and hence have been a very active area of research in the past decade. Yet the fine tuning of these devices at the molecular level, up to a few years ago, has been a relatively unexplored field. There are two classes of polymers used to fabricate organic photovoltaic (OPV) devices, main chain semi-conducting polymers which are conjugated through the backbone of the polymer and side chain semi-conducting polymers which are conjugated through the side chains of the polymer backbone.14
Main chain semi-conjugated polymers have high charge carrier mobilities which make them excellent transport materials. However, these materials are mainly hole-transporting (HT) and there are a more limited number electron transport materials (ET) such as oxadoles, triazoles, triazines and C60-fullerenes, often these materials have inherent difficulties associated with them, such as difficult monomer synthesis, low degrees of polymerization, broad polydispersities (PDI) and poor solution processability (compared to side chain counterparts).15 Due to the nature in which these polymers are synthesized it is not currently possible to chain extend hole transporting main chain polymers with electron transporting main chain polymers or vice versa. Devices therefore have to be fabricated by traditional blending techniques or by constructing a simple two layered device, thus potentially limiting their efficiency in OPV device applications due to the small interfacial area between the two heterojunctions. It is believed that an increase in the interface between the HT and ET materials would greatly enhance device performance as the interface is the active area for charge separation.16 As the exciton diffusion length is of the order of a few nanometres it is believed that such microphase separation could be achieved using block copolymers.
Poly(vinyltriphenylamine), TPA, is a well-established stable polymer with good hole conducting properties.17–20 TPA has been employed as a hole transporting building block for application in the synthesis of robust crosslinked materials for blue-emitting phosphorescent polymers and also in preparation of semi-conductor dendritic-linear block copolymers.21–23 There are many examples of the incorporation of triphenylamine (and its derivatives) units into the main chain of semi-conducting polymers using conventional coupling approaches.24–27 Research efforts have also focused upon the synthesis of side chain semi-conducting TPA polymers using recent advances in controlled radical polymerization (CRP). The interest in non-conjugated backbone polymers is due to their greater solubility compared to rigid rod polymers which in turn facilities their application in solution-processed devices. This follows on from the early work by Feast et al. using conventional radical techniques to polymerize p-TPA and more recent work by Chen and co-workers28–30 Indeed, recently there have been a number of reports of the application of polymers prepared by CRP techniques coupled with hole-conducting polymers, such as the work of Harth and co-workers using nitroxide mediated polymerization (NMP) techniques31 and by Ludwigs and co-workers for the preparation of mesoporous hole-conducting block copolymer templates.32 In addition, a recent report has used ring opening metathesis polymerization (ROMP) for the preparation of polynorborenes whose side chain is functionalized with TPA groups.33 Furthermore, the preparation of PTPA using living techniques was reported in 2009 by Higashihara and Ueda using sec-BuLi to afford well-defined polymers (Mw/Mnca. 1.1) which could be incorporated into PS and PMMA blocks and also form multiblocks with polythiophene.34 In addition, an alternative approach by the group of Thelakkat reported the post-polymerization modification using Pd coupling of a bromostyrene polymer prepared by atom transfer radical polymerization to afford PTPA.35
An early example of a semi-conducting diblock copolymer system was first reported by Behl et al. in 2002.36 In this work and in later reports they produced hole and electron conducting diblock copolymers using TEMPOvia a NMP mechanism. Vinyl-triphenylamine (TPA) units were used as the hole transporting (HT) layer and triphenyltriazine derivatives were used as the electron transporting (ET) layer.37 It is well documented that diblock copolymers can form various interesting nanostructured morphologies due to the phase separation of the incompatible blocks. It has been proposed that the fine control over the morphologies of semi-conducting diblock copolymers could enhance device performance.12 This has been shown to good effect by Thelakkat and co-workers who used NMP to synthesis well defined diblock copolymers (with Mw/Mn's below 1.5) containing a HT poly(p-TPA) segment and an ET poly(perylene acrylate) segment.38,39 It was shown that the system had one order of magnitude better photovoltaic device performance than the analogous copolymer blend which was attributed to its ability to nanophase separate into micro-domains of the same order as the exciton diffusion length. Fréchet and co-workers have also reported the use of NMP to synthesize such semi-conducting diblock copolymer systems again using TPA as the HT transporting moiety.40 However, it was reported that the para analogue of the TPA monomer (p-TPA) was unstable in storage, gave low polymer conversions and broad polydispersity indexes compared to the m-TPA analogue. Further work in 2007 from the Fréchet group described the preparation of HT-b-ET copolymers using NMP techniques in which the HT block was TPA.12 More recently, Natori et al. have reported the polymerization of p-TPAvia an anionic polymerization method and also reported the chain extension of these polymers with styrene.41 The use of NMP is reported in the literature as the main CRP methodology for the preparation of semi-conducting diblock copolymers, however NMP only allows for the polymerization of a relatively narrow class of monomers and does not readily allow α and ω chain end functionalization. One interesting exception to this was the report in 2006 by Lindner and Thelakkat of the synthesis of a perylene functionalized alkoxyamine and the subsequent polymerization of p-TPA monomers. The resultant well-defined polymers exhibited photoluminescence quenching due to the electron transfer between the donor polymer chain and the acceptor dye molecule on the α chain end.42 Both ATRP and RAFT allow for ready end group functionalization as has been demonstrated in 2008 by Zhu and co-workers, who incorporated a TPA group into a chain transfer agent for RAFT.43 The polymerization of p-TPA as a HT vinyl monomer has been demonstrated in the literature not only to proceed viaNMP but also through ATRP techniques, by Huck and Friend et al. in 2005 to grow semi-conducting polymers from surfaces to afford TPA polymer brushes.44,45 To date there are number of literature reports of the application of RAFT as a technique to achieve semi-conducting polymers yet there have only been a handful of reports of its application for the preparation of semi-conducting diblock copolymers. For example, the polymerization of N-vinyl carbazole, a well-established HT block, has been widely reported using xanthate mediated RAFT.46–48 Only very recently Zorn and Zentel reported the first use of RAFT polymerization for the synthesis of conducting polyTPA along with an anchoring block for binding to oxide semiconductors.49 The same authors along with the group of Char in 2009 demonstrated the application of these blocks for the preparation of quantum dot-block copolymers.50 A second report in 2009, from Theato and co-workers reported the RAFT polymerization of p-TPA and the preparation of blocks which can be modified post-polymerization to afford a second block for anchoring to oxide semiconductors.51
Since the advent of RAFT, it has been shown to be perhaps the most versatile of CRP methodologies and offers great control over a wider range of monomeric species in a variety of reaction conditions to yield polymers with predictable molecular weights and low polydispersity indexes.52–57 Another major advantage of RAFT is the facile synthesis of chain transfer agents (CTA) which allows for near infinite functionality to be incorporated into both the R and Z group positions to achieve bifunctional polymers.58–60 Perhaps more important is the ability to modify the thiocarbonyl Z group post polymerization to achieve a host of functional polymers such as via an aminolysis or reduction route to achieve thiol functionalized polymers.61Polymers with thiols on the chain end are of great interest in microelectronics due to their reactivity with semiconducting nanoparticles (NPs) such as gold. NPs are promising building blocks for conducting materials as they have tunable broad spectral absorptions that typically coincide with the visible region of the solar spectrum making them intriguing materials for use in OPV device applications. Therefore it is highly desirable to incorporate NPs with semi-conducting polymers therefore forming NP nanohybrids that could have inherit lightweight flexibility, mechanical strength, good solution processability, broad spectral absorption, good photostability, and high charge carrier mobility.62,63 The ability to chemically anchor a monolayer polymer coating directly onto NPs is key to ensuring solubility and miscibility of the NP and also gives a well controlled interface, which facilitates the efficient charge transfer and exciton separation. The ability to post functionalize RAFT polymers to obtain a thiol end functionalized polymer to chemically anchor to NPs is extremely valuable and has been demonstrated in the literature using non conducting polymers.64 McCormick and co-workers reported the in situreduction of the dithioester group on various non conducting polymers and copolymers in the presence of gold nanoparticles and their subsequent absorption to achieve inorganic hybrid materials.65
For the realization of precisely supramolecular assembled electronic devices the synthesis of well defined semi-conducting polymers by CRP techniques such as RAFT are of particular interest, as are the precise morphologies they may form with semi-conducting nanocrystals such as gold. In this work, we report the design and synthesis of a RAFT CTA to mediate the controlled polymerization of TPA and fluorinated TPA vinyl monomers to ultimately achieve semi-conducting block copolymers with tunable chain end functionalization. We also report initial results on the selective solution self-assembly of the diblocks into well-defined phase separated nanostructures. This method allow for the preparation of materials which incorporate the 3 features required for light absorption and hence represents a modular approach for the creation of nanostructures with a large interface and good electrooptical properties.
Experimental section
Materials
Unless otherwise stated, chemicals were used as received from Aldrich and used without further purification. AIBN (2,2′-azo-bis(isobutyronitrile)) was recrystallized twice from methanol and stored in the dark at 4 °C. Bromostyrenes were filtered over silica to remove inhibitor immediately prior to use. Dry toluene was obtained by passing over a column of activated alumina using an Innovative Technologies solvent purification system.Instrumentation
Nuclear magnetic resonance (NMR) experiments (1H and 13C) were performed on a Bruker AVANCE 400 FT-NMR spectrometer using deuterated solvents. Coupling constants are in Hertz, and chemical shifts are reported in parts per million relative to CHCl3 (7.26 ppm for 1H and 77.2 ppm for 13C). Extended 1H and 13C NMR spectra were recorded on an AVANCE 500 Cryo FT-NMR spectrometer, all at 25 °C. Size exclusion chromatography (SEC/GPC) measurements were performed using a Viscotek VE1122 solvent delivery system with a Viscotek VE3580 RI detection system using THF as eluent at a flow rate of 1 mL min−1. The molecular weights of polymers were calculated relative to ten polystyrene standards (Mp 580 to 377![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 Da) with 2 PLgel 5 μm mixed C columns (300 mm × 7.5 mm). Infrared spectroscopy was recorded on a Perkin Elmer, Spectrum 100 FT-IR Spectrometer. UV-vis spectra were recorded on a Varian Cary 4000 UV-vis spectrophotometer in tetrahydrofuran unless otherwise stated. Microanalyses were performed using a CE-440 Elemental Analyzer. DSC measurements were performed on a Mettler Toledo, HP DSC827 with analysis performed using Mettler Toledo STARe software v9.20. The samples were run at a heating rate of 10 °C min−1 and the Tgs were taken as the midpoint of the inflection tangent. Hydrodynamic diameters (Dh) and size distributions of the nanostructures in aqueous solutions were determined by dynamic light scattering (DLS). The DLS instrumentation consisted of a Malvern Zetasizer NanoS instrument operating at 25 °C or 65 °C with a 4 mW He–Ne 633 nm laser module. Measurements were made at a detection angle of 173° (back scattering), and Malvern DTS 5.02 software was used to analyze the data. Samples for TEM analysis were prepared by glow discharge followed by deposition of a solution of graphene oxide, allowed to air dry and then a solution of sample was deposited and allowed to dry. All samples were then examined with a transmission electron microscope (JEOL TEM-1200), operating at 100 kV. Samples for TEM analysis were prepared by glow discharge of a lacy carbon TEM grid followed by deposition of a solution of graphene oxide. Average sizes of the micelles were determined from counting the sizes of at least 100 particles for 3 different images. UPS data was collected on a photoemission yield spectroscope, Model AC-2 (Riken Keiki Co.,Ltd.) with an energy scan range of 4.6 to 6.2 eV, spot area 4 × 4 mm and variable UV intensity. Cyclic voltammtery was performed using 0.1 M tetrabutylammonium hexafluorophosphate dissolved in anhydrous dichloromethane as electrolyte solution. A platinum disk (0.02 cm2) and a platinum wire were used as working and counter electrodes, respectively, as well as Ag/AgCl in 3 M NaCl(aq) solution as a reference. Electroanalytical measurements were performed using a Gamry 600 potentiostat-galvanostat.
400 Da) with 2 PLgel 5 μm mixed C columns (300 mm × 7.5 mm). Infrared spectroscopy was recorded on a Perkin Elmer, Spectrum 100 FT-IR Spectrometer. UV-vis spectra were recorded on a Varian Cary 4000 UV-vis spectrophotometer in tetrahydrofuran unless otherwise stated. Microanalyses were performed using a CE-440 Elemental Analyzer. DSC measurements were performed on a Mettler Toledo, HP DSC827 with analysis performed using Mettler Toledo STARe software v9.20. The samples were run at a heating rate of 10 °C min−1 and the Tgs were taken as the midpoint of the inflection tangent. Hydrodynamic diameters (Dh) and size distributions of the nanostructures in aqueous solutions were determined by dynamic light scattering (DLS). The DLS instrumentation consisted of a Malvern Zetasizer NanoS instrument operating at 25 °C or 65 °C with a 4 mW He–Ne 633 nm laser module. Measurements were made at a detection angle of 173° (back scattering), and Malvern DTS 5.02 software was used to analyze the data. Samples for TEM analysis were prepared by glow discharge followed by deposition of a solution of graphene oxide, allowed to air dry and then a solution of sample was deposited and allowed to dry. All samples were then examined with a transmission electron microscope (JEOL TEM-1200), operating at 100 kV. Samples for TEM analysis were prepared by glow discharge of a lacy carbon TEM grid followed by deposition of a solution of graphene oxide. Average sizes of the micelles were determined from counting the sizes of at least 100 particles for 3 different images. UPS data was collected on a photoemission yield spectroscope, Model AC-2 (Riken Keiki Co.,Ltd.) with an energy scan range of 4.6 to 6.2 eV, spot area 4 × 4 mm and variable UV intensity. Cyclic voltammtery was performed using 0.1 M tetrabutylammonium hexafluorophosphate dissolved in anhydrous dichloromethane as electrolyte solution. A platinum disk (0.02 cm2) and a platinum wire were used as working and counter electrodes, respectively, as well as Ag/AgCl in 3 M NaCl(aq) solution as a reference. Electroanalytical measurements were performed using a Gamry 600 potentiostat-galvanostat.
Synthesis
:1 petroleum ether/ethyl acetate) to yield 1.31 g (86%) of 1 as a bright yellow viscous oil. Elem. Anal.: Found C, 49.27; H, 4.95; C10H12OS3 expected C, 49.14; H, 4.95; IRνmax/cm−1 3315–2950 (broad), 2919, 2877, 1494, 1452, 1053, 1002, 797, 660; 1H NMR (CDCl3) δ = 2.38 (1H, br s, CH2OH), 3.66 (2H, t, S–CH2–CH2), 3.91 (2H, t, S–CH2–CH2), 4.69 (2H, s, CH2–Ph), 7.28–7.40 (5H, m, Ph); 13C NMR (CDCl3) δ = 39.8, 42.3, 61.2, 128.5, 129.9, 135.4, 224.1.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 5.55 (1H, d, CHHCH), 6.60 (1H, dd, Ar–CHCH2), 7.01 (3H, m, ArH), 7.09 (5H, m, ArH), 7.25 (6H, dd, ArH). 13C NMR (CDCl3) δ = 114.3, 120.7, 122.4, 122.9, 124.0, 124.4, 129.5, 129.6, 136.8, 138.9, 147.9, 148.3.
CH), 5.55 (1H d, CHHCH), 6.60 (1H, dd, Ar–CHCH2), 7.01 (3H, m, ArH), 7.09 (5H, m, ArH), 7.25 (6H, dd, ArH). 13C NMR (CDCl3) δ = 114.3, 120.7, 122.4, 122.9, 124.0, 124.4, 129.5, 129.6, 136.8, 138.9, 147.9, 148.3.
CH), 5.65 (1H, d, CHHCH), 5.90 (1H, s, NH), 6.60 (1H, dd, Ar–CHCH2), 6.95 (1H, d, ArH), 7.10 (2H, m, ArH), 7.25 (4H, m, ArH). 13C NMR (CDCl3) δ = 113.0, 114.5, 116.0, 118.0, 119.5, 122.5, 123.0, 124.5, 130.0, 136.0, 139.0, 141.0.
CH), 5.65 (1H, d, CHHCH), 6.55 (1H, dd, Ar–CHCH2), 6.90 (1H, d, ArH), 7.10 (1H, s, ArH), 7.25 (2H, m, ArH), 7.40 (4H, s, 4H ArH), 7.45 (2H, s, ArH). 13C NMR (CDCl3) δ = 116.0, 117.0, 118.5, 121.5, 123.0, 124.0–126.0, 127.5, 131.5, 133.0–135.0, 136.5, 141.0, 145.5, 147.5.
CH), 5.55 (1H, d, CHHCH), 6.60 (1H, dd, Ar–CHCH2), 7.01 (3H, m, ArH), 7.09 (5H, m, ArH), 7.25 (6H, dd, ArH). 13C NMR (CDCl3) δ = 114.3, 120.7, 122.4, 122.9, 124.0, 124.4, 129.5, 129.6, 136.8, 138.9, 147.9, 148.3.
CH), 5.55 (1H d, CHHCH), 6.60 (1H, dd, Ar–CHCH2), 7.01 (3H, m, ArH), 7.09 (5H, m, ArH), 7.25 (6H, dd, ArH). 13C NMR (CDCl3) δ = 114.3, 120.7, 122.4, 122.9, 124.0, 124.4, 129.5, 129.6, 136.8, 138.9, 147.9, 148.3.
CH), 5.65 (1H, d, CHHCH), 5.90 (1H, s, NH), 6.60 (1H, dd, Ar–CHCH2), 6.95 (1H, d, ArH), 7.10 (2H, m, ArH), 7.25 (4H, m, ArH). 13C NMR (CDCl3) δ = 113.0, 114.5, 116.0, 118.0, 119.5, 122.5, 123.0, 124.5, 130.0, 136.0, 139.0, 141.0.
CH), 5.65 (1H, d, CHHCH), 6.55 (1H, dd, Ar–CHCH2), 6.90 (1H, d, ArH), 7.10 (1H, s, ArH), 7.25 (2H, m, ArH), 7.40 (4H, s, 4H ArH), 7.45 (2H, s, ArH). 13C NMR (CDCl3) δ = 116.0, 117.0, 118.5, 121.5, 123.0, 124.0–126.0, 127.5, 131.5, 133.0–135.0, 136.5, 141.0, 145.5, 147.5.
Homopolymers 7–13. The polymer solution was precipitated into diethyl ether to remove monomer and filtered from the solvent over a fritted funnel (×3), to afford white and yellow polymers that were dried in a vacuum oven overnight at 40 °C. Percentage conversion was calculated by comparing monomer vinyl units at 5.10 and 5.55 ppm with polymer backbone units at 0.75–2.45 ppm and polymer aromatic side chain units at 5.80–7.20 ppm. End group analysis using 1H NMR spectroscopy was used to calculate Mn by comparing the pyrene signals from the CTA at 7.85–8.35 ppm with polymer backbone units at 0.75–2.40 ppm and polymer aromatic side chain units at 5.80–7.20 ppm assuming 100% Z group fidelity. DSC: 13 – Tg = 119 °C and Tm = 150 °C.
Homopolymer 14. The polymer solution was completely dried from solvent on high vacuum line and washed with CHCl3 to remove monomer over a fritted funnel (×3), to afford a yellow polymer that was dried in a vacuum oven overnight at 40 °C. Percentage conversion was calculated from 1H NMR by comparing integrals of monomer vinyl signals at 5.20 and 5.65 ppm with polymer backbone signals at 0.75–2.45 ppm and polymer aromatic side chain signals at 5.50–7.65 ppm. End group analysis using 1H NMR spectroscopy was used to calculate Mn by comparing the pyrene signals from the CTA at 7.70–8.25 ppm with polymer alkyl units at 0.75–2.40 ppm and polymer aromatic side chain units at 5.50–7.65 ppm assuming 100% Z group fidelity. DSC: 14 – Tg = 126 °C and Tm = 161 °C.
Chain extended polymers 15, 16, 17, 19 and 20. The polymer solution was precipitated into diethyl ether and filtered from the solvent over a fritted funnel (×3), to remove monomer and afford white and yellow polymers that were dried in a vacuum oven at 40 °C.
Block copolymers 18 and 21. The polymer solution was completely dried on a high vacuum line and washed with petroleum ether over a fritted funnel (×3), to remove monomer and afford a yellow polymer that was dried in a vacuum oven at 40 °C. Percentage conversion was calculated from 1H NMR by comparing integrals of monomer vinyl signals at 5.20 and 5.65 ppm with polymer aromatic side chain signals at 7.20–7.65 ppm. End group analysis using 1H NMR spectroscopy was used to calculate Mn by comparing the pyrene signals from CTA 2 at 7.70–8.25 ppm with polymer aromatic side chain units at 5.50–7.65 ppm assuming 100% Z group fidelity.
Results and discussion
CTA
For the synthesis of well defined semi-conducting diblock copolymers we first needed to prepare a RAFT chain transfer agent (CTA) to mediate the polymerization of the monomers in a controlled manner. As both monomers that we will be using are styrenic derivatives there were two possible CTAs of choice, trithiocarbonates and dithioesters as both of these have been reported to polymerize styrenic derivatives with predictable molecular weights and low polydispersities.60 However, due to the harsh nature of Grignard chemistries used to synthesize dithioesters, we chose to use a trithiocarbonate as their facile synthesis allows for high yielding products in short reaction times and also allows functionality to be incorporated easily into both the R and Z group positions giving end functionalized polymers.61 A benzyl unit was chosen for the R group as this initiates and reinitiates readily to allow for the well controlled polymerizations of styrenic derivatives (Scheme S1†).67 As both the monomers that we synthesized have a broad UV absorption (ca. 0–500 nm), determination of end group fidelity or degree of polymerization by the trithiocarbonate group or any other chromophore is not possible. Instead we chose to incorporate pyrene into the Z group as this afforded distinct signals in the 1H NMR spectra completely separated from the homopolymers and the diblock copolymer aromatic signals in the spectra. This allowed the degree of polymerization to be calculated by end group analysis and also assessment of the polymer end group fidelity. Therefore, we synthesized CTA, 2 to mediate the polymerization of both monomers to ultimately produce a semi-conducting diblock copolymer with fine control over polydispersities and end group fidelity. Using previously reported facile chemistries, 1 was synthesized by reacting mercaptoethanol with carbon disulfide and benzyl bromide in the presence of potassium phosphate in THF.59 The product, 1 was isolated as yellow oil after flash chromatography in an overall yield of 86% and found to be pure by 1H and 13C NMR spectroscopies and elemental analyses. CTA 1 was then used in the synthesis of 2, by reacting it with pyrene butyric acidvia an esterification reaction. The product, 2 was separated from starting materials by flash chromatography as a yellow solid in a 60% yield and found to be pure by 1H and 13C NMR spectroscopies and elemental analyses (Fig. S1†).Monomers
N-vinylcarbazole is known to be a hole transporting material and has been shown to be polymerized via the RAFT mechanism with good control.68,69 However, its polymerizationvia RAFT can only be readily mediated using xanthates as the chain transfer agent. Therefore N-vinylcarbazole and styrenic derivatives are incompatible monomeric species and cannot be polymerized through the same RAFT center, preventing the facile synthesis of NVC (HT)-b-styrenic (ET) diblock copolymers. The literature shows that one of the most efficient and widely used HT species for the synthesis of semi-conducting materials are triphenylamine derivatives, and the p-vinyltriphenylamine (p-TPA) analogue has been reported for its use in CRP techniques and in particular with NMP, and more recently with ATRP and RAFT. However, it was suggested by Fréchet and co-workers that the p-TPA monomer showed limited stability compared to the m-TPA derivative.40 Thus, in this work both the p-TPA 3 and m-TPA 4 were synthesized as previously reported in the literature in 80% and 91% yields respectively by a Buchwald–Hartwig cross coupling reaction (see Scheme S2†).As we were interested in preparing diblock polymers we also needed to explore the RAFT polymerization of an ET monomer. Behl and Zentel have reported that the introduction of fluorinated groups, especially –CF3, which are so strongly electron withdrawing they can transform a typical HT material into an ET material by increasing the electron affinity of the material.37,70 Thus we designed and synthesized two new potential ET monomers, 5 and 6 modeled on the HT TPA monomer, by incorporating –CF3groups onto the phenyl groups. By using modified literature procedures, the synthesis of 6 was achieved in two steps (Scheme S3†). Firstly, bis(trifluoromethyl)aniline was reacted with 3-bromostyrene by a Buchwald–Harwig cross coupling reaction to give monomer precursor 5. The product 5 was isolated as a dark yellow/brown oil after flash chromatography in an overall yield of 70% and found to be pure by 1H and 13C NMR spectroscopies. Monomer 5 was then reacted with bis(trifluoromethyl)bromobenzene once again by a Buchwald–Hartwig cross coupling reaction to give monomer 6. The product 6 was isolated as white solid after flash chromatography in a yield of 98% and found to be pure by 1H and 13C NMR spectroscopy (Fig. 1).
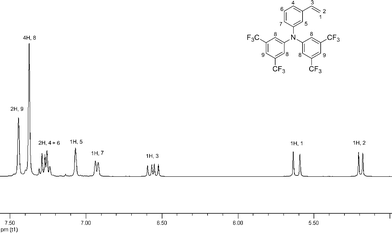 | ||
| Fig. 1 1H NMR spectra of 6 in CDCl3. | ||
Homopolymers and copolymers
The newly synthesized monomers were polymerized using CTA 2 as the initiator. The polymerizations were run in either 1,4-dioxane or neat at various reaction times, temperatures and concentrations of AIBN, except in the case of 14, where α,α,α-trifluorotoluene (TFT) was used due to the limited solubility of the polymers in non-fluorinated solvents at high conversions (Scheme 1). | ||
| Scheme 1 Conditions: (i) polymerization at 110 °C with CTA 2 and no AIBN; (ii) polymerization at 110 °C with no CTA 2 and no AIBN; (iii) polymerization at 90 °C with CTA 2 and AIBN. | ||
The molecular weights (MWs) of the polymers were determined by gel permeation chromatography (GPC) using polystyrene standards and nuclear magnetic resonance spectroscopy (NMR). It was found that for most polymers the theoretical MWs calculated from % conversion as determined by 1H NMR spectroscopy (by comparing monomer vinyl units (5.10 and 5.55 ppm for HT and 5.20 and 5.65 ppm for the fluorinated derivative) with polymer alkyl units (0.75–2.45 ppm for both) and polymer aromatic side chain units (5.80–7.20 ppm for HT and 5.50–7.65 ppm for the fluorinated derivative)), matched the MWs calculated by end group analysis using 1H NMR (by comparing the pyrene signals from the CTA (7.85–8.35 ppm) with polymer alkyl units (0.75–2.40 ppm) and polymer aromatic side chain units (5.80–7.20 ppm for HT and 5.50–7.65 ppm for the fluorinated derivative)). While GPC data obtained using polystyrene standards underestimated the MWs of all the polymers (Table 1 and 2), this is in agreement by previous reports by Fréchet.40
| Polymer | Monomer | Relative equivalentsa | T/°C | Time (mins) | % conv.e | M n e/kDa | M n g /kDa | M w/Mng |
|---|---|---|---|---|---|---|---|---|
|
a Ratio of [monomer]:[2]:[AIBN].
b
Polymerizations carried out in v/v 2:1 1,4-dioxane:monomer.
c
Polymerizations carried out in v/v 2:1 α,α,α-trifluorotoluene:monomer.
d
Polymerization carried out in the presence of AIBN and no CTA.
e Calculated by 1H NMR in CDCl3.
f Calculated by 1H NMR in d8-THF.
g Samples were measured by GPC (THF) using polystyrene standards.
|
||||||||
| 7 | 3 | [50]:[1]:[0]b |
110 | 60 | 80 | 11.4 | 6.8 | 1.32 |
| 8 | 4 | [50]:[1]:[0]b |
110 | 60 | 75 | 10.7 | 6.3 | 1.29 |
| 9 | 4 | [100]:[1]:[0]b |
110 | 35 | 49 | 13.8 | 9.5 | 1.19 |
| 10 | 3 | —d | 110 | 60 | 47 | — | 90.0 | 1.63 |
| 11 | 4 | —d | 110 | 60 | 41 | — | 92.0 | 1.51 |
| 12 | 3 | [50]:[1]:[0.1]b |
90 | 300 | 75 | 10.7 | 7.0 | 1.18 |
| 13 | 4 | [50]:[1]:[0.1]b |
90 | 300 | 76 | 10.8 | 7.5 | 1.17 |
| 14 | 6 | [50]:[1]:[0]c |
110 | 1800 | 75f | 21.4 | 10.6 | 1.16 |
| Polymer | Initiating polymer/monomer | Relative Equivalentsa | Temp./°C | Time (min) | % Conv. d | M n/kDae | M w/Mne |
|---|---|---|---|---|---|---|---|
|
a Ratio of [monomer]:[initiating polymer]:[AIBN].
b
Polymerizations carried out in v/v 2:1 1,4-dioxane:monomer.
c
Polymerizations carried out in v/v 3:3:1 άάά-trifluorotoluene:1,4-dioxane:monomer.
d Calculated by 1H NMR in d8-THF.
e Samples were measured by GPC (THF) using polystyrene standards.
|
|||||||
| 15 | 7/3b | [100]:[1]:[0] |
110 | 60 | — | 7.3 | 1.60 |
| 16 | 8/4b | [100]:[1]:[0] |
110 | 60 | — | 13.8 | 1.55 |
| 17 | 9/4b | [150]:[1]:[0] |
110 | 60 | — | 19.0 | 1.57 |
| 18 | 8/6c | [50]:[1]:[0] |
110 | 1200 | 80 | 9.3 | 1.37 |
| 19 | 12/3b | [100]:[1]:[0.1] |
90 | 300 | — | 14.2 | 1.23 |
| 20 | 13/4b | [100]:[1]:[0.1] |
90 | 300 | — | 15.3 | 1.20 |
| 21 | 13/6c | [50]:[1]:[0.1] |
90 | 1440 | >99 | 14.0 | 1.36 |
Homopolymers synthesized using vinyltriphenyl amine monomers 3 (polymer 7) and 4 (polymer 8) at 110 °C showed relatively broad polydispersity indexes (ca. Mw/Mn = 1.32 and 1.29 respectively) for a RAFT mediated polymerization suggesting it may have proceeded via a relatively uncontrolled mechanism. In addition, the chain extended polymers prepared from macroinitiators 7 and 8 with monomers 3 (polymer 15) and 4 (polymer 16) respectively confirmed that the polymerization of monomers 3 and 4 at 110 °C was indeed uncontrolled (Scheme 2, Table 2). Polymer 15 showed little or no noticeable chain extension in the overlay of GPC traces with starting polymer 7 and no increase in molecular weight by GPC (ca. Mn = 6.8 kDa for 7, and 7.3 kDa for 15), yet its polydispersity had broadened significantly (to ca. Mw/Mn = 1.60). The absence of yellow color in polymer 7 after precipitation is indicative that there is little or no trithiocarbonate end groups present in the polymer and was proven by elemental analyses which reported no sulfur content. This indicates that the polymerization did not proceed though a controlled RAFT mechanism but through an uncontrolled radical polymerization. However, polymer 16 showed noticeable chain extension in the overlay of GPC traces with starting polymer 8 and a significant increase in Mn by GPC (ca. Mn = 6.3 kDa for 8, and 13.8 kDa for 16), yet there was still a low weight molecular shoulder present which gave a broad polydispersity (ca. Mn/Mw = 1.55) (Fig. S5†).
 | ||
| Scheme 2 Conditions: (i) polymerization at 110 °C with CTA 2 and no AIBN; (ii) polymerization at 90 °C with CTA 2 and AIBN. | ||
This indicated that not all of homopolymer 8 had reinitiated upon chain extension and the presence of a low molecular shoulder suggested that polymer 8 had lost end group fidelity or similar to polymer 7, some of monomer 4 had also proceeded through an uncontrolled radical polymerization. This difference in polymerization properties between para and meta TPA analogues maybe due to electronic effects of the para (monomer 3) and meta (monomer 4) positions of the vinyl group and the ability of the meta and para substituents to stabilize the propagating radical formed.71 To confirm that the polymerization of monomers 3 and 4 was occurring through an uncontrolled mechanism via thermal initiation of the styrenic monomers and not through a controlled RAFT mechanism, the polymerization of monomers 3 and 4 were carried out in the absence of CTA 2. The data showed that the polymerization of these monomers could indeed be thermally initiated at 110 °C to afford polymers 10 and 11 respectively with relatively broad polydispersity indexes (ca. Mw/Mn = 1.63 and 1.51 respectively).
The RAFT polymerizations using monomer 6 (to afford polymer 14) at 110 °C gave high molecular weight homopolymers with relatively low polydispersity indexes indicating that the polymerization had proceeded through a controlled RAFT mechanism (ca. Mn = 21.4 kDa and Mw/Mn = 1.16). Diblock copolymer 18 synthesized from the chain extension of macroinitiator 8 with monomer 6, showed promising chain extension data (Fig. 2c), although it was expected that macroinitiator 8 could not completely chain extend from the data obtained for the chain extension to form polymer 16. When overlaying the GPC trace of polymer 18 with that of polymer 8 one can see an increase in Mn (ca. Mn = 6.3 kDa for 8, and 9.3 kDa for 18), showing that the polymerization of monomer 6 is compatible with monomer 4 and that the synthesis of diblock copolymers are achievable using RAFT.
 | ||
| Fig. 2 (a) GPC traces of p-TPA homopolymer/macroinitiator 12 and p-TPA chain extended polymer 19 showing complete chain extension of the starting polymer (b) GPC traces of m-TPA homopolymer/macroinitiator 13 and m-TPA chain extended 20 showing complete chain extension of starting polymer. (c) GPC traces of m-TPA homopolymer/macroinitiator 8 and block copolymer 18 with initiating polymer shoulder. (d) GPC traces of m-TPA homopolymer/macroinitiator 13 and chain extended polymer 21 showing complete chain extension of the starting polymer. | ||
To prevent thermal initiation of styrenic monomers 3 and 4 from occurring and to obtain well defined homopolymers and block copolymers with high end group fidelity via RAFT, polymerizations of monomers 3 and 4 with CTA 2 were carried out in the presence of a radical initiator, AIBN at 90 °C. Homopolymers synthesized using vinyltriphenyl amine monomers 3 (polymer 12) and 4 (polymer 13) at 90 °C showed relatively low polydispersity indexes indicating that the polymerization had proceeded through a controlled RAFT mechanism (ca. Mw/Mn = 1.18 for 12 and 1.17 for 13) (Table 1). The chain extended polymers prepared from macroinitiators 12 and 13 with monomers 3 (polymer 19) and 4 (polymer 20) respectively confirmed that the polymerization of monomers 3 and 4 at 90 °C were indeed controlled (Table 2). Polymer 19 showed complete chain extension in the overlay of GPC traces with starting polymer 12 (Fig. 2a) and a significant increase in Mn by GPC (ca. Mn = 7.0 kDa for 12, and 14.2 kDa for 19), with a slight broadening in polydispersity (ca. Mw/Mn = 1.23) (Table 2).
Further exploration of the kinetics of the polymerization of p-TPA to form polymer 12 indicated that the kinetic plot displays semi-linear kinetics indicating that the polymerization may not be completely controlled (Fig. 3a). This may also be illustrated in the plot of molecular weight [Mn(GPC)] against % conversion (NMR) for polymer 12 where although there is a steady increase in Mn there is a large broadening in the polydispersity of the intermediate polymers (Fig. 3b). The m-TPA chain extension polymer 20 also showed good chain extension in the overlay of GPC traces with starting polymer 13 (Fig. 2b) and a significant increase in Mn by GPC (ca. Mn = 7.5 kDa for 13, and 15.3 kDa for 20), with slight broadening in polydispersity (ca. Mw/Mn = 1.20). The kinetic plot for the m-TPA polymer 13 displays pseudo first order kinetics indicative of controlled radical polymerization (Fig. 3c). The controlled nature of the polymerization of monomer 4 is also be illustrated in the plot of molecular weight [Mn(GPC)] against % conversion (NMR) for polymer 13 where there is a steady increase in Mn with narrow polydispersity indexes of the intermediate polymers (Fig. 3d). Although it was possible to chain extend polymer 12, given the slightly broader polydispersity indexes and problematic kinetic data obtained for monomer 3, and its reported difficulties in handling and storage,40 we subsequently chose monomer 4 (m-TPA) for further polymerizations to form the diblock copolymers.
![(a) A kinetic plot for the RAFT polymerization of HT monomer 3 with CTA 2 at 90 °C in 1,4-dioxane 2 : 1 w/monomer. [M] : [CTA] : [AIBN] = [50] : [1] : [0.1]. (b) A plot of molecular weight [Mn(GPC)] against % conversion (NMR) of HT monomer 3 with CTA 2; the solid line is Mn,theor(NMR), with polydispersities. (c) A kinetic plot for the RAFT polymerization of HT monomer 4 with CTA 2 at 90 °C in 1,4-dioxane 2 : 1 w/monomer. [M] : [CTA] : [AIBN] = [50] : [1] : [0.1]. (d) A plot of the molecular weight [Mn(GPC)] against % conversion (NMR) of HT monomer 4 with CTA 2; the solid line is Mn,theor(NMR), with polydispersities.](/image/article/2011/PY/c0py00359j/c0py00359j-f3.gif) | ||
| Fig. 3 (a) A kinetic plot for the RAFT polymerization of HT monomer 3 with CTA 2 at 90 °C in 1,4-dioxane 2:1 w/monomer. [M]:[CTA]:[AIBN] = [50]:[1]:[0.1]. (b) A plot of molecular weight [Mn(GPC)] against % conversion (NMR) of HT monomer 3 with CTA 2; the solid line is Mn,theor(NMR), with polydispersities. (c) A kinetic plot for the RAFT polymerization of HT monomer 4 with CTA 2 at 90 °C in 1,4-dioxane 2:1 w/monomer. [M]:[CTA]:[AIBN] = [50]:[1]:[0.1]. (d) A plot of the molecular weight [Mn(GPC)] against % conversion (NMR) of HT monomer 4 with CTA 2; the solid line is Mn,theor(NMR), with polydispersities. | ||
The chain extension of macroinitiator 13 (m-TPA) with monomer 6 at 90 °C gave the proposed diblock copolymer 21. When comparing the GPC traces of polymer 21 with polymer 13 a significant increase in molecular weight can be observed indicating that chain extension has occurred, although the trace from polymer 21 has broadened significantly and still overlaps with the trace from starting polymer 13 which would indicate that not all of the starting polymer may have chain extended (Fig. 2d). However, this may be an artifact of the GPC as the fluorinated groups may be interacting with the GPC column and account for the broadening in polydispersity (ca. Mw/Mn = 1.36 for 21 and 1.17 for 13). Analysis of the extended 1H NMR spectrum of this diblock confirms an almost 1:1 degree of polymerization for both blocks (HT29-b-fluorinated TPA35). However, when considering the mass fraction of this polymer the mass ratios are closer to 1:2 given the higher mass of the fluorinated block relative to the PTPA block. The block copolymer 21 is thermally stable with Tonsets obtained from TGA well above 300 °C. DSC measurements showed that the glass transition temperatures of the block copolymers are in the same range as the homopolymers (13 – 119 °C and 14 – 126 °C). This data alongside the solution assembly data indicates microphase separation of the diblock occurs.
Self-assembly
Using this diblock copolymer, 21, the self-assembly behavior was explored. Initially we performed a solvent screen to assist in the choice of solvents for self assembly. These results indicated that the PTPA block was soluble in polar solvents such as chloroform, dioxane and THF whilst the fluorinated block was not soluble in either of the first 2 of these solvents but was soluble in TFT and THF. Hence, THF was chosen as a good solvent for both blocks and when the diblock was dissolved at loadings from 1 to 10 mg mL−1 only small 3–4 nm sized particles were observed by DLS analysis. However upon added 9 volume equivalents of chloroform to a solution of 21 in THF, to give a final concentration of 1 mg mL−1, large particles were observed by DLS analysis (22, Dh = 60 nm, PDI 0.12). A similar experiment was performed by adding 9 volume equivalents of TFT, which is a non-solvent for the PTPA block, to a solution of 21. In this case much larger ill-defined particles were observed by DLS analysis (23, Dh = 214 nm, PDI 0.40). To further explore the morphology of the well-defined nanostructures of sample 22TEM analysis without staining was performed (Fig. 4). From these experiments it was clear that spherical micellar morphologies (Dav = 56 ± 4 nm) formed with the particles of 22 having the fluorinated block in the core (evidenced from the high contrast of the fluorine groups on a graphene oxide support which allowed for facile imaging without staining). | ||
| Fig. 4 A TEM image of the self-assembled particles 22. | ||
Electronic properties
Cyclic voltammetry (CV) measurements (as described by Behl and Zentel) were used to confirm the properties of the new monomer 6.37 In these experiments monomer 6 showed a reduction potential at ca. −1 V yet in the oxidation direction no activity was observed. Further analysis of the polymers was performed by UV-vis spectroscopy in THF. For polymers 12, 13 and 14 the resultant UV-vis spectra showed broad absorptions around 300 nm. For each polymer the absorption cut-off was utilized to calculate the optical band gap energy (Egopt, Table 3). For polymer 14, which was the fluorinated TPA derivative a smaller energy band gap was calculated compared to the unfluorinated derivatives indicating stabilization of the LUMO upon fluorination. This data was utilized alongside CV measurements (of a thin polymer film) to calculate the HOMO and LUMO levels for each polymer. The values for 12 and 13 compare well with previous literature reports for similar polymers prepared by other methods.21,30 Indeed we also observed reversible electrochemistry for the triphenylamine polymers with no para-substitution as reported by Fréchet and coworkers.40 CV measurements of the polymer 14 showed significant reduction activity compared to the unfluorinated derivative 13 as well as less pronounced oxidation activity as expected upon the introduction of CF3 groups,37 this confirms the increased electron conducting properties of 14 (see ESI†). Furthermore, UPS was used to confirm the HOMO level for polymers 13 and 14, which were both ca. −5.61 eV. Further CV measurements of films of the block copolymer 21 indicate that both the reduction of the fluorinated and the oxidation of the PTPA occur at nearly unchanged potential (Eox = 1.01 and 0.86 V) compared to the homopolymers suggesting that nanophase separation of the blocks was occurring.| Polymer | Reduction potential (V vs. FOC)a | Oxidation potential (V vs. FOC)a | E g opt/eVb | HOMOc (eV vs. vacuum) | LUMOd (eV vs. vacuum) |
|---|---|---|---|---|---|
| a Reduction potentials were measured by cyclic voltammetry. b Optical bandgaps were calculated from the absorption spectra of the polymer solutions. c HOMO levels were estimated from the LUMO level (determined by CV) and the optical energy gaps. d LUMO levels were converted from measured reduction potentials by CV (ELUMO = EHOMO + Egopt) assuming the absolute energy level of ferrocene to be 4.8 eV.72 | |||||
| 12 | −0.99 | 1.14 | 3.53 | −5.94 | −2.41 |
| 13 | −0.99 | 1.12 | 3.50 | −5.92 | −2.42 |
| 14 | −0.99 | 0.95 | 3.30 | −5.75 | −2.45 |
Conclusions
Using a trithiocarbonate chain transfer agent we have been able to demonstrate the controlled polymerization of homo and diblocks of a triphenylamine and a fluorinated triphenylamine monomer. The resultant homo and diblock copolymers are of interest due to their hole or electron transporting abilities and hence these new diblocks may be of interest in the preparation of devices. In addition, this methodology allows for the ready incorporation of a functional group (in this case pyrene) at the polymer chain end. The ability to introduce functionality into the polymer chain end allows for the potential application of these materials to interface within inorganic materials. For example in this work the pyrene chain end functionalized polymers may find application in the functionalization of carbon nanotubes.73 Overall, the application of facile RAFT polymerization technologies has been demonstrated to allow for the preparation of materials of potential interest in optoelectronic applications. Further work expanding the class of monomers polymerizable by this method is underway to allow for the facile tuning of the HOMO and LUMO levels of the two blocks to enable the ready tuning of polymer properties.Acknowledgements
The authors thank Professor Sir Richard Friend (University of Cambridge) for valuable discussions, the mass spectrometry service (University of Warwick) for their invaluable help, Dr G. Pace for help with CV analysis (Optoelectronics Group, University of Cambridge), Dr Jun Oguma Visiting Scientist in the Optoelectronics Group (University of Cambridge) for assistance with UPS. Dr Neil Wilson, Department of Physics, Warwick is thanked for assistance with TEM measurements. The authors thank the EPSRC, the Royal Society, Emmanuel College Cambridge, IRC in Nanotechnology (University of Cambridge) and the University of Warwick for funding. Some of the equipment used in this research was obtained, through Birmingham Science City with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF).Notes and references
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591–2611 CrossRef CAS.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581–2590 CrossRef CAS.
- H. Shirakawa, Angew. Chem., Int. Ed., 2001, 40, 2574–2580 CrossRef.
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121–127 CrossRef CAS.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- J. Kido, K. Nagai, Y. Okamoto and T. Stotheim, Appl. Phys. Lett., 1991, 59, 2760–2762 CrossRef CAS.
- F. Garnier, Chem. Phys., 1998, 227, 253–262 CrossRef CAS.
- A. R. Murphy, J. Liu, C. Luscombe, D. Kavulak, J. M. J. Fréchet, R. J. Kline and M. D. McGehee, Chem. Mater., 2005, 17, 4892–4899 CrossRef CAS.
- R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205–9216 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS.
- B. Ma, B. J. Kim, L. Deng, D. A. Poulsen, M. E. Thompson and J. M. J. Fréchet, Macromolecules, 2007, 40, 8156–8161 CrossRef.
- N. Haberkorn, M. C. Lechmann, B. H. Sohn, K. Char, J. S. Gutmann and P. Theato, Macromol. Rapid Commun., 2009, 30, 1146–1166 CrossRef CAS.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. E. Newman, J. Quin, F. Dolz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- M. Stoclka, D. M. Pai, D. S. Renfer and J. F. Yanus, J. Polym. Sci., Part A: Polym. Chem., 1983, 21, 969–983 Search PubMed.
- M. Tamada, H. Koshikawa, F. Hosoi, T. Suwa, H. Usui, A. Kosada and H. Sato, Polymer, 1999, 40, 3061–3067 CrossRef CAS.
- M. Redecker, D. D. C. Bradley, M. Inbasekaran, W. W. Wu and E. P. Woo, Adv. Mater., 1999, 102, 241–245 CrossRef.
- J. P. Chen, D. Markiewicz, V. Y. Lee, G. Klaerner, R. D. Miller and J. C. Scott, Synth. Met., 1999, 107, 203–207 CrossRef CAS.
- A. S. Lang, F. R. Kogler, M. Sommer, U. Wiesner and M. Thelakkat, Macromol. Rapid Commun., 2009, 30, 1243–1248 CrossRef CAS.
- M. S. Liu, Y. H. Niu, J. W. Ka, H. L. Yip, F. Hunag, J. Luo, T. D. Kim and A. K. Y. Jen, Macromolecules, 2008, 41, 9570–9580 CrossRef CAS.
- X. Wang, Z. Chen, K. Ogino, H. Sato, K. Strzelec, S. Miyata, Y. Luo and H. Tan, Macromol. Chem. Phys., 2002, 203, 739–747 CrossRef CAS.
- A. Bolognesi, P. Betti, S. Destri, U. Giovanella, J. Moreau, M. Pasini and W. Porzio, Macromol. Rapid Commun., 2009, 42, 1107–1113 CAS.
- S. Maria, A. S. Sushsa, M. Sommer, D. V. Talapin, A. L. Rogach and M. Thelakkat, Macromolecules, 2008, 41, 6081–6088 CrossRef CAS.
- W. Zhang, Z. Fang, M. Su, M. Saeys and B. Liu, Macromol. Rapid Commun., 2009, 30, 1533–1537 CrossRef CAS.
- Z. G. Zhang, K. L. Zhang, C. X. Zhu, K. G. Neoh and E. T. Kang, Macromolecules, 2009, 42, 3104–3111 CrossRef CAS.
- W. J. Feast, R. J. Peace, I. C. Sage and E. L. Wood, Polymer, 1999, 42, 167–174.
- K.-M. Yeh, C.-C. Lee and Y. Chen, Synth. Met., 2008, 158, 565–571 CrossRef CAS.
- C.-C. Lee, K.-M. Yeh and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7960–7971 CrossRef CAS.
- C. T. Adkins, H. Muchalski and E. Harth, Macromolecules, 2009, 42, 5786–5792 CrossRef CAS.
- E. J. W. Crossland, P. Cunha, S. Scroggins, S. Moratti, O. Yurchenko, U. Steiner, M. Hillmyer and S. Ludwigs, ACS Nano, 2010, 4, 962–966 CrossRef CAS.
- J. H. Park, C. Yun, M. H. Park, Y. Do, S. Yoo and M. H. Lee, Macromolecules, 2009, 42, 6840–6843 CrossRef CAS.
- T. Higashihara and M. Ueda, Macromolecules, 2009, 42, 8794–8800 CrossRef CAS.
- K. Peter and M. Thelakkat, Macromolecules, 2003, 36, 1779–1785 CrossRef CAS.
- M. Behl, E. Hattemer, M. Brehmer and R. Zentel, Macromol. Chem. Phys., 2002, 203, 503–510 CrossRef CAS.
- M. Behl and R. Zentel, Macromol. Chem. Phys., 2004, 205, 1633–1643 CrossRef CAS.
- G. Krausch and M. Thelekkat, Angew. Chem., Int. Ed., 2006, 45, 3364–3368 CrossRef CAS.
- S. M. Lindner and M. Thelakkat, Macromolecules, 2004, 37, 8832–8835 CrossRef CAS.
- L. Deng, P. T. Furuta, S. Garon, J. Li, D. Kavulak, M. E. Thompson and J. M. J. Fréchet, Chem. Mater., 2006, 18, 386–395 CrossRef CAS.
- I. Natori, S. Natori, H. Usui and H. Sato, Macromolecules, 2008, 41, 3852–3858 CrossRef CAS.
- S. M. Lindner and M. Thelakkat, Macromol. Chem. Phys., 2006, 207, 2084–2092 CrossRef CAS.
- J. Fu, Z. Zhang, Z. Cheng, J. Zhu, W. Zhang and X. Zhu, Polym. Bull., 2008, 61, 287–297 CrossRef CAS.
- H. J. Snaith, G. L. Whiting, B. Sun, N. C. Greenham, W. T. S. Huck and R. H. Friend, Nano Lett., 2005, 5, 1653–1657 CrossRef CAS.
- G. L. Whiting, H. J. Snaith, S. Khodabakhsh, J. W. Andreasen, D. W. Bereiby, M. M. Nielsen, N. C. Greenham, R. H. Friend and W. T. S. Huck, Nano Lett., 2006, 6, 573–578 CrossRef CAS.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS.
- W. Y. Tam, C. S. K. Mak, A. M. C. Ng, A. B. Djurisic and W. K. Chan, Macromol. Rapid Commun., 2009, 30, 622–626 CrossRef CAS.
- H. Mori, H. Ookuma, S. Nakano and T. Endo, Macromol. Chem. Phys., 2008, 207, 1005–1017.
- M. Zorn and R. Zentel, Macromol. Chem. Phys., 2008, 29, 922–927 CrossRef CAS.
- M. Zorn, W. K. Bae, J. Kwak, H. Lee, C. Lee, R. Zentel and K. Char, ACS Nano, 2009, 3, 1063–1068 CrossRef CAS.
- D. Kessler, M. C. Lechmann, S. Noh, R. Berger, C. Lee, J. S. Gutmann and P. Theato, Macromol. Chem. Phys., 2009, 30, 1238–1242 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, Aust. J. Chem., 2006, 59, 661–662 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- G. Moad, M. Chen, M. Haussler, A. Postma, E. Rizzardo and S. H. Thang, Polym. Chem., 2011 10.1039/c0py00179a.
- M. R. Wood, D. J. Duncalf, S. P. Rannard and S. Perrier, Org. Lett., 2006, 8, 553–556 CrossRef CAS.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 4183–4185 RSC.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- G. Schmidt and M. M. Malwitz, Curr. Opin. Colloid Interface Sci., 2003, 8, 103 CrossRef CAS.
- Z. Lin, Chem.–Eur. J., 2008, 14, 6294–6301 CrossRef CAS.
- B. J. Kim, S. Given-Beck, J. Bang, C. J. Hawker and E. J. Kramer, Macromolecules, 2007, 40, 1796–1798 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS.
- A. C. Evans, J. Skey, M. Wright, W. J. Qu, C. Ondeck, D. A. Longbottom and R. K. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6814–6826 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- H. Mori, S. Nakano and T. Endo, Macromolecules, 2005, 38, 8192–8201 CrossRef CAS.
- H. Mori, H. Ookuma, S. Nakano and T. Endo, Macromol. Chem. Phys., 2006, 207, 1005–1017 CrossRef CAS.
- A. Facchetti, M. Mushrush, H. E. Katz and T. J. Marks, Adv. Mater., 2003, 15, 33–38 CAS.
- K. Harata, K. Nagata, T. Hasegawa and H. Yuki, Makromol. Chem., 1977, 178, 2413–2419 CrossRef.
- C. C. Wu, J. C. Strum, R. A. Register, J. Tian, E. P. Dana and M. E. Thompson, IEEE Trans. Electron Devices, 1997, 44, 1269–1281 CrossRef CAS.
- S. Meuer, L. Braun and R. Zentel, Macromol. Chem. Phys., 2009, 210, 1528–1535 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Further synthetic details and characterization data for the polymers and micelles. See DOI: 10.1039/c0py00359j |
| ‡ Present address: Department of Chemical Engineering and Material Science, University of Minnesota, Minneapolis, USA. |
| This journal is © The Royal Society of Chemistry 2011 |