Recent advances in entropy-driven ring-opening polymerizations
Satu
Strandman
a,
Julien E.
Gautrot
b and
X. X.
Zhu
*a
aDépartement de chimie, University of Montreal, CP 6128 Succursale Centre-ville, Montreal, Quebec H3C 3J7, Canada. E-mail: sstrandman@gmail.com; julian.zhu@umontreal.ca; Fax: +1 514 340 5290; Tel: +1 514 340 5172
bDepartment of Chemistry, Melville Laboratory for Polymer Synthesis, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: juliengautrot@yahoo.fr; Fax: +44 1223 334 866; Tel: +44 1223 336 401
First published on 1st December 2010
Abstract
Entropy-driven ring-opening polymerization (ED-ROP) of unstrained macrocyclic monomers and/or oligomers employs the ring-chain equilibria between macrocycles and their corresponding polymers and the associated increase of conformational freedom to achieve high molecular weight materials. The principles of building macrocyclic compounds, their use in ED-ROP, and the practical considerations of polymerizations are described, and recent progress in this area is discussed through selected examples. The various polymerization techniques used for ED-ROP are discussed, including anionic, radical, coordination/insertion, ring-opening metathesis, and enzymatic polymerization methods. Emphasis is placed on the potential of ED-ROP in the synthesis of biomaterials and the development of enzyme-catalyzed green systems.
 Satu Strandman | Satu Strandman conducted her studies at the University of Helsinki, Finland at the Laboratory of Polymer Chemistry with Professor H. Tenhu and received her PhD in 2008 on amphiphilic star block copolymers. After her PhD, she worked in the research group of Professor F. Winnik at the Université de Montréal, Canada, with alkynylpyrenes and alkynylpyrene-functionalized polymers. Recently, she has joined the research group of Professor X. X. Zhu. Her research interests include synthesis and characterization of complex macromolecular architectures, stimuli-responsive polymers, and biocompatible polymeric systems. |
 Julien E. Gautrot | Julien Gautrot received his PhD from the University of Manchester, Department of Chemistry, in 2004, under the supervision of Professor P. Hodge, designing novel bioinspired conjugated organic materials. He then worked as a postdoctoral researcher with Professor X. X. Zhu, at the Université de Montréal, Canada, on the synthesis of novel degradable materials based on bile acids. In 2007, he moved back to the UK, where he currently works with Professor W. Huck, University of Cambridge, Melville Laboratory, on the development of novel biofunctional coatings and the study of the cell–matrix interface. |
 X.X. Zhu | X. X. Zhu received his BSc degree in chemistry from Nankai University in China, and his PhD degree from McGill University in Canada. After postdoctoral work at CNAM, France and the University of Toronto, he joined the Chemistry Department of Université de Montréal in 1992, where he is now a professor and holds the Canada Research Chair in polymeric biomaterials. He and his group make use of natural compounds such as bile acids in the preparation of polymers for use in biomedical and pharmaceutical applications. He is author of over 170 research publications and several patents and book chapters. |
1. Introduction
Ring-opening polymerization (ROP) is a fundamental method of polymer synthesis and has been employed extensively for small ring monomers (5–8 atoms) to produce polymers such as aliphatic polyesters, polyamides, and polycarbonates, among others.1 The polymerization of low-molar mass rings is driven by the relief in ring strain, an enthalpy-driven process. When the ring size is large enough (usually ≥14 atoms), changes in enthalpy upon opening are minimal and polymerization becomes entropy-driven (ED-ROP) through an increase of conformational freedom.2Although the ED-ROP itself is neither a step-growth nor a chain-growth process,3 its background lies in the step-growth polymerizations which produce a fraction of cyclic oligomers for statistical reasons. The presence of cyclics may significantly alter the properties of polymeric product if their fraction is large, i.e., if the synthesis is carried out in high dilution. For example, cyclic poly(ethylene terephthalate) oligomers can migrate on the surface of spun fibres and interfere with their dyeing.2 When the polymerization is carried out neat, the amount of cyclics can be lower than 2 weight percent of the product.4 Entropy-driven ROP exploits a ring-chain equilibrium between macrocycles and polymer chains, which is adjustable by altering the concentration of the reaction system. High dilution favors the monomers or oligomers (macrocycles of varying size), while high concentration favors the polymeric product (Fig. 1). The equilibrium nature of entropy-driven ROP leads to the “most probable distribution” of molar masses, Mw/Mn ≈ 2.0.3
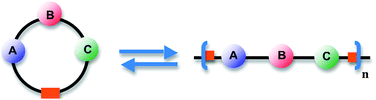 | ||
| Fig. 1 The ring-chain equilibrium. | ||
ED-ROP has been successfully applied to a number of systems and it can be carried out via several polymerization mechanisms such as anionic,5,6 insertion/coordination,7 radical,8 enzymatic,9 or ring-opening metathesis polymerization (ROMP).10 ROMP of small rings by well-defined catalysts exhibits living character allowing the synthesis of uniform polymers and block copolymers.11 Despite its non-living nature, ED-ROP of large macrocycles displays unique advantages. The large number of atoms constituting the macrocyclic backbone ensures the great diversity of polymeric structures providing a cornucopia of unexplored macromolecular systems. The use of macrocycles allows their functionalization in positions that do not interfere with the ring-opening activity and hence nearly any chemical moiety can be inserted in the polymer main chain for optimizing the physical properties and degradation of materials as well as for introducing functionalities for molecular recognition, photoluminescence, supramolecular interactions, and biocompatibility.
Recently, a review has been published on ED-ROP with a focus on applications of the resulting materials.12 In the current review article, we wish to emphasize the development of different polymerization methods and mechanisms of ED-ROP and discuss recent advances in this field for the design of green polymerization systems.
2. Principles of ED-ROP
2.1. Building macrocycles
The synthesis of macrocyclic compounds has attracted a great deal of interest due to their importance in organic and natural compound synthesis as well as in supramolecular and organometallic chemistry. Macrocycles are also common in antitumoral, antibiotic, and antifungal compounds.13 More recently, the potential of macrocyclic compounds in materials synthesis and engineering has been discovered. In general, cyclic compounds fall into three categories: (a) strained small rings with kinetically favored synthesis (n ≤ 7,14n = number of atoms in ring); (b) strained medium rings with less kinetically favored formation (n = 8–13 reported for lactones,14n = 8–11 for cycloolefins15); and (c) kinetically unfavorable strainless large rings (n ≥ 14 reported for lactones,14n ≥ 12 for cycloolefins15). The competition between the intramolecular reaction (cyclization) and intermolecular reaction (polymerization) brings additional challenge to the synthesis of macrocycles.The macrocyclization is highly dependent on the flexibility of starting compounds.16 An example of this can be taken from the synthesis of bile acid-based cyclic monomers. Bile acids are a class of steroids with a rigid bent shape of the steroidal backbone (Scheme 1). The rigidity of backbone predisposes the molecules to form large oligomeric macrocycles in three major distributions. If spacers between the steroid ring and the reactive site are sufficiently long and flexible, cyclic monomers prevail. When slightly smaller and/or more rigid spacers are incorporated, cyclic dimers are the main product. When the linkers are absent or too rigid, trimers and tetramers are the main cyclic oligomers.16
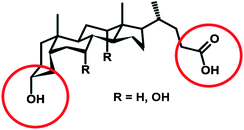 | ||
| Scheme 1 General structure of bile acids. The common linker-bearing groups are circled. | ||
So far, the majority of large-ring macrocycles used in ED-ROP has been carboxylic acid esters,10,17 carbonates,17 amides,18 alkenes,19 aromatic ether ketones,6b or sulfones.20 A classical strategy for building macrocycles involves a reaction of α,ω-difunctional compound(s) under high dilution to favor cyclization over polymerization. If macrocycles are not under dynamic equilibrium with their oligomers and polymers in the reaction conditions, the so-called pseudo-high dilution techniques can be applied.21 In a small-scale synthesis of cyclic oligo-depsipeptides or oligoesters, oligomers can be built on a polymer support, and upon cyclization by di-n-butyltin oxide only cyclic species are released in solution in moderate to good yields.22,23
Ring-closing metathesis (RCM) has gained popularity in building cyclic alkene derivatives owing to its moderate tolerance to functional groups24 which facilitates inserting complex moieties such as bile acids,25,26 calix[4]arenes,27 or 1,10-phenanthrolines28 in the macrocycle. This method provides cyclization of bisalkenes by ruthenium-based catalysts in nonpolar solvents such as toluene or dichloromethane, which favors the initiation of metathesis reaction.29 Recently, a thorough review has been published on the equilibrium nature of ring-closing metathesis reactions.15
A different approach on the preparation of macrocycles employs ring-chain equilibrium in a reverse way: cyclic species are formed by depolymerizing macromolecules under high dilution in the presence of appropriate catalyst or enzyme. The process is called cyclodepolymerization (CDP) or ring-closing depolymerization (RC-DP). The first examples of CDP date back to 1930s on aliphatic polyesters30 and polycarbonates,31 after which the scope of polymers has been expanded to polyamides,32 polyurethanes,33 high-performance polyethersulfones,34,35 and polyetherketones.36 In some of these examples cyclics can be removed by distillation, which makes high dilution unnecessary. More recent strategies for CDPs have exploited ring-closing metathesis on alkene-containing aliphatics, polyesters and polyamides,10,17,18,37 or enzymatic degradation of polyesters.9b,38
2.2. Theory behind ED-ROP
The theoretical background of ED-ROP lies in the pioneering work of Jakobson and Stockmeyer39 on intramolecular reaction in polycondensations. This theory describes the distribution of cyclic and linear polymers at equilibrium in concentrated solutions and predicts a critical monomer concentration [M]c, below which the condensing system can be converted entirely into rings. Since enthalpic effects are neglected and the rings are assumed to be strainless, the theory overestimates the equilibrium constant for cyclization and hence the critical monomer concentration for smaller cyclics.Later, Kornfield and co-workers40 improved the prediction of these factors by introducing an enthalpy change due to ring strain energy and proposing a revised critical monomer concentration [M]c,∞, which can be used for predicting the polymerizability of cyclic monomers at a given temperature. If the initial monomer concentration exceeds this critical concentration, most of the additional monomers contribute to polymerization.
When enthalpy change is not the driving factor of polymerization, polymerization conditions need to be such that entropic changes become significant. The negative and concentration-dependent translational entropy is high for small rings and becomes smaller upon increasing ring size. The positive rotational and torsional entropies decrease much less upon increasing the ring size and can become dominant over the translational entropy in large rings.41 Ultimately, the positive entropic contribution upon conformational flexibility of the polymer chain can become prevalent, thus driving the polymerization of large macrocycles.15 In a dilute system, the translational entropy of the monomeric unit is higher and the equilibrium is shifted towards the cyclics.
2.3. Practical considerations
As already emphasized, concentration is a crucial factor for ring-opening polymerizations. While macrocyclizations and cyclodepolymerizations are carried out in a millimolar (mM) range of concentrations,13,42 ED-ROP is typically carried out in 0.1–5.0 M solutions or even in the neat.10 If solvent-free conditions are employed, the polymerization should be conducted above the glass transition temperature (Tg) or melting temperature (Tm) of the polymer and monomer.3 In general, temperatures for conducting ED-ROP in solution are low (typically ≤50 °C, depending on the solvent). No excess heat or volatiles are released during the entropy-driven polymerization as the large macrocycles are strainless and the ring-opening involves only shuffling of linkages between the repeating units.The effect of solvent, however, goes beyond the concentration as the solvent quality is expected to influence the extent of backbiting (cyclization) reactions. A higher value of critical monomer concentration [M]c is predicted for a thermodynamically good solvent.43 The solvent has also been reported to affect the equilibrium E/Z ratios of metathesis reactions,37 but other factors such as the type of catalyst and reaction time have also been studied.12,44 Finally, the solvent influences the activity of a metathesis catalyst: the initiation rates of Grubbs' 1st generation catalyst have been reported to be proportional to the dielectric constant of nonpolar solvents.29
The choice of catalyst is important in ring-opening metathesis polymerization (ROMP) and ED-ROMP reactions, where high activity and solubility need to be combined with functional group tolerance. Since their discovery in the late 1990s, Grubbs' Ru-based catalysts, particularly the 2nd generation catalyst,45 have become highly popular owing to their robustness and commercial availability.11,44 Despite being less sensitive to oxygen or water, the susceptibility of the catalyst to coordinating agents remains a problem.13,15 Also the removal of metals from final product is challenging, putting pressure towards developing polymer or inorganic-support immobilized and recyclable catalysts. For this purpose, a variety of polymers have been investigated and summarized in more detail in recent reviews.46–48 As an example, an amphiphilic block copolymer has been synthesized by Bergbreiter and co-workers by ROMP using polyisobutylene (PIB) bound ruthenium-based metathesis catalyst. The ligand's PIB chain provided the end group of product polymer chain, resulting in block copolymerization.49
Another strategy for reducing the amounts of trace metals is using enzymatic polymerization. Enzymatic ring-opening polymerization has been utilized in the synthesis of polyesters, polycarbonates, polyphosphates, polythioesters, and poly(ester-amides).50–52 This method is particularly suitable for ED-ROPs as the polymerization can be conducted in bulk, reaction conditions are mild, and high molar masses are obtained from the polymerization of macrolides (large ring lactones) with relative ease.50 Higher rates of lipase-catalyzed polymerization of macrolides have been reported in comparison to low-molar mass lactones, which has been assigned to the higher hydrophobicity of macrolides promoting lactone–lipase complex formation53 or to the conformation-based accessibility of ester group in the enzyme's active site.54 However, comparison of kinetic parameters of polymerization of macrolides of various sizes catalyzed by Pseudomonas fluorescens lipase suggested that the increased polymerizability of macrolides would stem rather from the large ring size than better binding ability.55 More detailed discussion on the mechanism and kinetics of lipase-catalyzed ROP can be found elsewhere.52
Finally, supramolecular chemistry has brought an intriguing contribution on ED-ROP by building noncovalent polymers through hydrogen bonding. Depending on concentration, ureidopyrimidinone derivatives formed cyclic dimers or linear polymers with very high degree of polymerization (DP = 3200).56 The ring-chain equilibrium shifted toward linear chains at higher temperatures. This represents a special case of ED-ROP, because no catalyst or initiator is needed and no covalent bonds are formed.
3. Polymerization techniques compatible with ED-ROP
3.1. Anionic polymerization
Anionic ring-opening polymerization is commonly used in synthesizing polyesters from lactones. The polymerization of large lactone rings occurs by the attack of an anion on carbonyl carbon atom leading to acyl-oxygen scission and formation of an alkoxide growing species (Scheme 2).57 ED-ROP of 12- and 13-membered lactones was carried out by Endo and co-workers5 either in bulk or in THF using sodium, lithium or potassium methoxides as initiators at various temperatures (0–120 °C), resulting in polyesters with good yields. Number-average molar masses (Mn) of the polyesters were 3400–13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1. The ring-strain energy was equal for 12- and 13-membered rings, but the rate of propagation was higher for the larger ring, which the authors assigned to the difference in s-character of carbon atomic orbitals on the basis of NMR coupling constants.5
700 g mol−1. The ring-strain energy was equal for 12- and 13-membered rings, but the rate of propagation was higher for the larger ring, which the authors assigned to the difference in s-character of carbon atomic orbitals on the basis of NMR coupling constants.5
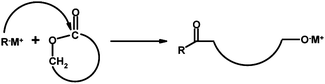 | ||
| Scheme 2 Initiation of anionic polymerization of large-ring lactones.57 | ||
Other groups of macrocycles polymerizable via anionic mechanism include ether ketones,58,59 ether sulfones20 and aromatic thioethers,60 although the latter ones undergo also thermally initiated free radical polymerization.8a,c,61 The anionic polymerization of cyclic ethers and their derivatives is initiated by CsF or alkali phenoxides. The phenoxides are considered more efficient than fluorides, and the efficiency order of the counterions is Cs > K > Na.20 Highly active catalysts often give polymers with very high molar masses and/or some branching, and thus with limited solubility.6b,20,36 In a recent example of the anionic method by Chen and co-workers,59 macrocyclic aryl ketone oligomers were synthesized via modified Friedel-Crafts acylation, and polymerized by anionic ED-ROP catalyzed with potassium 4,4′-biphenoxide in melt, yielding thermostable poly(ether ketones and sulfones) (Scheme 3) with poor solubility in common organic solvents, an indication of high molar masses. The melt viscosities at the initial stage of polymerization were low and increased slowly as the cyclic oligomers acted as a “lubricant” between the polymer chains.59
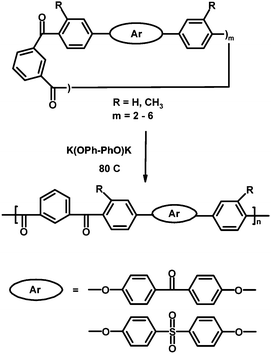 | ||
| Scheme 3 Synthesis of poly(ether ketones and sulfones) via anionic ED-ROP, K(OPh–PhO)K = potassium 4,4′-biphenoxide.59 | ||
3.2. Radical polymerization
The advantages of thermally initiated entropy-driven free radical ring-opening polymerization lay in the absence of added catalyst, and in the possibility for reactive molding while avoiding the problems in processing high melt viscosity polymers. For instance, nonisothermal heating of low-viscosity macrocyclic arylene thioether ketones with porous alumina membranes produced flexible polymeric microfibrils and microtubules of 200–400 nm in diameter (Fig. 2), and free-standing structures were obtained after removal of the membrane in alkaline conditions.8b Similarly, adhesive polydisulfide films were prepared from cocyclic arylene disulfide oligomers over thin aluminium plates.62 In catalytic reactive molding, initiator and monomer are mixed prior to polymerization, or the membrane can be impregnated with the initiator such as CsF for anionic polymerization, thus confining the polymerization within the pores.63 | ||
| Fig. 2 Poly(arylene thioether ketone) microfibrils synthesized within a porous alumina membrane8b (reprinted with permission from ref. 8b, Copyright 2010 American Chemical Society). | ||
3.3. Coordination/insertion polymerization
In addition to anionic polymerization, polyesters are often synthesized via so-called pseudoanionic or coordination/insertion ring-opening polymerization. In this method, the propagation proceeds by coordination of the monomer to active species, followed by insertion of the monomer into metal–oxygen bond. The growing chain remains attached to the metal during the propagation.64 The most widely used initiators are aluminium and tin alkoxides and carboxylates, and among them the most popular ones are tin(II) ethylhexanoate also known as stannous octoate, Sn(Oct)2, and di-n-butyltin oxide, SnO(Bu)2.57A recent high-throughput application of ED-ROP by Hodge and co-workers65 with coordination/insertion mechanism has utilized macrocyclic oligoesters (Scheme 4) for producing a library of polyesters and copolyesters with variable monomer ratios and molar masses up to 28300 g mol−1 (Mw). The syntheses were catalyzed by di-n-butyltin oxide in bulk at small scale (90 mg of monomers). The method worked well for all the other esters but phenolic ones, which was thought to be a result of the nature of the catalyst or unsuitable reaction conditions. The initial products were nonrandom copolymers as verified by 13C NMR spectroscopy, but longer reaction times resulted in random copolymers.65
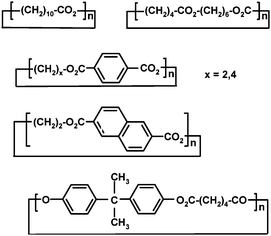 | ||
| Scheme 4 Macrocyclic oligomers for copolymerization by coordination/insertion mechanism.65 | ||
While ED-ROP allows introducing main-chain functionalities, it may also provide control over the sequence of repeating units when they are included in the same macrocycle. An example of such control using coordination/insertion mechanism is the work by Tolman and co-workers,7c who prepared an isomerically pure 14-membered cyclic diester to synthesize isotactic polymers with perfectly alternating lactic acid and alkylene oxide subunits (Scheme 5) upon ED-ROP by zinc alkoxide in toluene at room temperature. The monomer/catalyst ratio controlled linearly the molar masses at the range of 4900–72000 g mol−1 (Mn), but some backbiting reactions were observed at high conversions. The alternating polymers were completely miscible with atactic polylactide, which allowed tuning of the glass transition temperature of the blends.7c
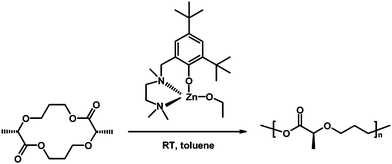 | ||
| Scheme 5 Synthesis of stereoregular poly(3-methyl-1,4-dioxane-2-one) by coordination/insertion ED-ROP.7c | ||
3.4. Ring-opening metathesis polymerization
Advances in catalyst development have made ring-opening metathesis polymerization (ROMP) an important class of ROP reactions. As ring-closing metathesis is convenient for building macrocyclic compounds with various functional components, their polymerization via ED-ROMP has become a promising method for building novel advanced materials. Recently, a detailed review has highlighted the scope of side chain functionalized materials that are synthesized mainly by enthalpy-driven ROMP of norbornene derivatives.66In ROMP, a transition metal (typically Ru or Mo) complex coordinates to a cyclic olefin double bond and subsequent [2 + 2]-cycloaddition gives a four-membered metalla-cyclobutane intermediate which will further undergo a cycloreversion to afford a new metal alkylidene, now larger in size. In the propagation stage, analogous steps are repeated until termination occurs. The propagating centers of the polymer may exist either in metallacyclobutane or metal alkylidene forms and the polymerization is reversible, i.e. ring-chain equilibrium prevails.11
The first studies of modern ED-ROMPs with Ru-based catalysts were conducted by Grubbs and co-workers67 with cyclic ethers. In comparison to acyclic diene metathesis polymerization of α,ω-dienes (ADMET), ED-ROMP proved to be more efficient for the formation of high-molar mass products. The highest molar mass obtained by Grubbs and Maynard through ED-ROMP was 206000 g mol−1 (Mn).67b Hodge and Kamau10 expanded the ring sizes of macrocyclic olefinic esters to 21–84-membered rings, which polymerized fast upon the evaporation of solvent: molar masses up to 94000 g mol−1 (Mn) were obtained in 10 minutes. Using olefin-containing cyclic oligoamides was more problematic due to the poor solubility of amides and their polymers.18 In addition, macrocyclization of amides by ring-closing metathesis (RCM) resulted in the deactivation of the catalyst when an amide group was in close proximity to the reactive double bond. Nevertheless, ED-ROMP of cyclic oligoamides was conducted successfully in solution (THF at 56 °C) and the solubility was improved by copolymerization with cyclic oligoesters.18
Some incidental observations of the RCM syntheses,68,69 as well as more detailed knowledge of factors influencing the ring-chain equilibria15 have inspired researchers to incorporate more complex functional moieties in cyclic compounds to yield main chain functionalized polymers. Yang and Swager27 synthesized main-chain calix[4]arene elastomers by copolymerizing calixarene-derived olefinic macrocycles with cyclooctene and norbornene (Scheme 6). The highest molar mass of a copolymer was 209000 g mol−1 (Mn) with a monomer ratio 1:5:2, for the respective monomers. The conformation of calixarenes influenced directly the mechanical properties of corresponding polymers. The more flexible calixarene building blocks provided higher mechanical strength and toughness by facilitating greater polymer–polymer interactions.27 An interesting example of ED-ROMP from Mayer and co-workers involves the polymerization of olefinic [2]catenanes to yield polypseudorotaxanes with phenanthroline ligands (Scheme 7).28 Threaded macrocycles were held in the backbone through copper–bis-phenanthroline complexes and released upon a demetallation treatment of polypseudorotaxane with KCN. The bare backbone had a molar mass of 93000 g mol−1 (Mw) corresponding to a degree of polymerization of 63.28
![Calix[4]arene-based macrocycles and their copolymerization.27](/image/article/2011/PY/c0py00328j/c0py00328j-s6.gif) | ||
| Scheme 6 Calix[4]arene-based macrocycles and their copolymerization.27 | ||
![ED-ROMP of [2]catenane for polypseudorotaxanes; n = 4.28](/image/article/2011/PY/c0py00328j/c0py00328j-s7.gif) | ||
| Scheme 7 ED-ROMP of [2]catenane for polypseudorotaxanes; n = 4.28 | ||
Gross and co-workers polymerized double bond-bearing natural lactonic sophorolipids by three different Ru catalysts, which gave polymers with Mn ≥ 42200 g mol−1 in good yields (≥67%) in 5 min at 25 °C.70 Sophorolipids are microbial glycolipid biosurfactants with a wide range of potential therapeutic applications.71 The lactonic sophorolipids with cis double bond were separated from a mixture of linear and lactonic forms produced by yeast Candida bombicola.72 The obtained poly(sophorolipids) (Scheme 8) comprised of a mixture of cis and trans isomers.70 The semicrystalline polysophorolipids are expected to find their applications as bioresorbable materials.73
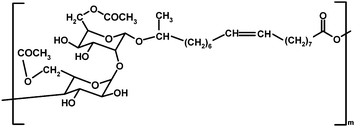 | ||
| Scheme 8 Chemical structure of a poly(sophorolipid).73 | ||
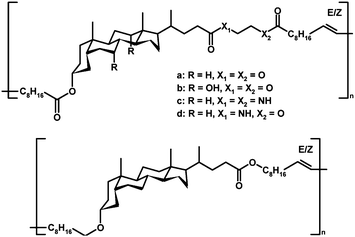
Another group of natural compound-based main-chain functionalized polymers was introduced by Gautrot, Zhu and co-workers, who have prepared macrocyclic bile acid-based olefinic esters and amides (Scheme 9) yielding amorphous thermoplastics with outstanding thermally activated shape memory properties (demonstrated in Fig. 3) and tunable mechanical behavior.25,26,74,75 While ADMET of a diene precursor of cyclic bile acid ester afforded polymers with a typical molar mass value of 22300 g mol−1 (Mn),26 ED-ROMP of the corresponding cyclic monomer yielded molar masses up to 152000 g mol−1 (Mn).26 All polymers were amorphous, as indicated by the transparency of solvent-casted films and verified by X-ray scattering experiments.75 The shape memory properties, i.e. strain fixity and strain recovery, which describe the ability of chain segments to fix the mechanical deformation and the ability of the material to memorize its permanent shape,76 were among the best reported for uncrosslinked amorphous polymers.74 Such properties were assigned to ordered domains acting as pseudo-crosslinks helping in freezing the transient shape due to increased intermolecular interactions, which depended on the number of hydroxyl groups of the bile acid moieties.75 Both the mechanical properties and glass transition temperatures (Tg) were tuned by copolymerization of the macrocycles with another cyclic monomer, ricinoleide, derived from castor oil.26,74 These features provide an opportunity for designing natural compound-based biomaterials with controllable mechanical and chemical properties as well as biocompatibility and bioresponsiveness.
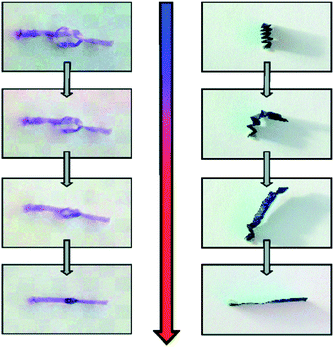 | ||
| Fig. 3 Demonstrating the shape recovery of bile acid main chain polymer films (reprinted with permission from ref. 74, Copyright 2010 American Chemical Society).74 | ||
3.5. Enzymatic polymerization
Increasing environmental concerns have turned researchers to explore greener alternatives for materials synthesis. Enzymes have been utilized as a non-toxic alternative for metal catalysts since the beginning of the 1990s, and particularly lipases have been studied in the syntheses and hydrolyses of polyesters and polycarbonates.52,77,78 The active site of lipases is –CH2OH of the serine residue, and lipase-catalyzed reactions proceed via an acyl–enzyme intermediate, enzyme-activated monomer (EM, Scheme 10).79 As discussed above, an enzymatic approach has been shown to be particularly suitable for ED-ROP of macrolides, such as 11-undecanolide,80 12-dodecanolide,80 15-pentadecanolide,81 and 16-hexadecanolide55 (12–17-membered rings). Heise and co-workers have demonstrated the biocompatibility of polymers of 15–17-membered lactones synthesized by an enzymatic method.82 Lipase-catalyzed ROP can be carried out in bulk or in organic solvents (e.g. toluene, heptane, or 1,4-dioxane) but also greener alternatives (e.g. water, ionic liquids, or supercritical carbon dioxide—scCO2) have often been employed.52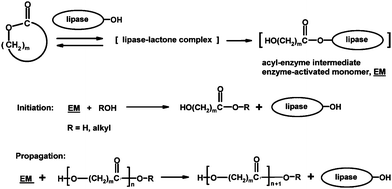 | ||
| Scheme 10 Principle of lipase-catalyzed ring-opening polymerization.79 | ||
Enzyme-catalyzed ED-ROP of cyclic oligomers has been considered as a simple and green method towards high molar mass diol–diacid polyesters, such as poly(butylene adipates) and poly(butylene succinates).9b,38 Direct enzyme-catalyzed polycondensation of adipic acid and butane-1,4-diol has been successful, but molar masses have generally been low and the removal of condensation product methanol is difficult, which can be avoided by the ring-opening polymerization.38,83 In addition, large ring monomers and oligomers for ED-ROP can be produced either by enzymatic cyclization reactions or by lipase-catalyzed cyclodepolymerization of synthetic or microbial polyesters thus enabling recycling of polymers (Scheme 11).9b,84–86
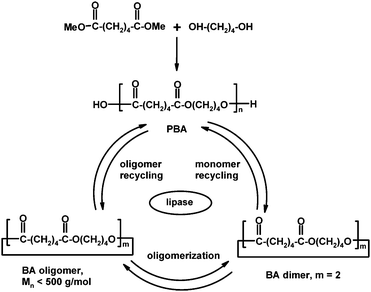
Recently, Hodge and co-workers87 explored the scope of polymer-supported Candida antarctica (CA) lipase B in ED-ROP of 12–84-membered macrocycles bearing lithocholic acid moieties. Although CA lipase-catalyzed oligocondensation of cholic acid was reported earlier,88 Hodge and co-workers did not detect any polymer in the case of unsubstituted lithocholic acid or its cyclic dimers and trimers. However, the lipase-catalyzed ED-ROP of larger lithocholic acid-based macrocycles in anhydrous toluene at 70 °C gave polymers and copolymers (Scheme 12) in high yields, and molar masses of 9100–25400 g mol−1 (Mn).87 This suggests that even large cyclics are accessible to the active site of the enzyme.
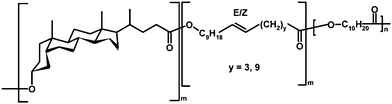 | ||
| Scheme 12 Copolymer of a bile-acid based macrocycle by lipase-catalyzed ED-ROP.87 | ||
4. Future perspectives and concluding remarks
To summarize, entropy-driven ring-opening polymerizations exploiting the well-known phenomenon of ring-chain equilibrium can be carried out by various mechanisms to yield high-molar-mass main-chain functionalized polymers and copolymers. In recent years, studies on ED-ROP have focused on the development of novel functional materials as well as environmentally friendly processes and products. The advantages of ED-ROP lie in its lack of byproducts or released heat, and the possibility of carrying out the synthesis under solvent-free conditions. Since the reaction is reversible, cyclodepolymerization can be employed for recycling of polymers by different mechanisms and continuous enzyme-catalyzed processes have already been developed for recycling of polyesters.51 The production of macrocycles, however, requires large amounts of solvents, whether they are produced by ring-closing reactions or by cyclodepolymerization, and a great deal of effort is put on developing ring-closing and ROP processes in “green solvents” such as water, ionic liquids or supercritical CO2, particularly in enzymatic catalysis.51,66As metathesis reactions proceed via double bonds of olefins, processing of compounds from renewable resources such as vegetable oils could provide means towards environmentally friendly products.24 Metathesis catalysts are still expensive and need developing for further synthetic control over ring-opening polymerizations. In addition, the potential toxicity of catalysts and their difficult removal bring serious limitations for food industry and biomedical applications. Enzymatic methods are a highly promising alternative for polymerizing macrocyclic compounds in the absence of metal catalysts, some of which are difficult to polymerize by chemical means.50 Although the first steps have already been taken towards chemical versatility through enzymatic ED-ROP, the obtained molar masses are still relatively low and the reactions proceed slowly.
So far, the range of polymers and materials produced by ED-ROP has been encouraging, from polyethers and high-performance polymers to polypseudorotaxanes and shape-memory elastomers. Without doubt this scope will expand hand-in-hand with further advances in enthalpy-driven ring-opening polymerizations.
Acknowledgements
We wish to acknowledge the Natural Sciences and Engineering Research Council (NSERC) of Canada, Fonds Québecois de Recherche sur la Nature et les Technologies (FQRNT) and the Canada Research Chair program for financial support. We are members of Centre for Self-Assembled Chemical Structures (CSACS) funded by FQRNT and Groupe de Recherche en Sciences et Technologies Biomédicales (GRSTB) funded by FRSQ. S.S. thanks FQRNT for a postdoctoral scholarship (Programme de Bourses d'Excellence pour Étudiants Étrangers).References
- (a) G. Odian, Principles of Polymerization, John Wiley & sons, Hoboken, 4th edn, 2004 Search PubMed; (b) Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier, and J. M. Raquez, Wiley-VCH Verlag GmbH & Co., Weinheim, 2009 Search PubMed.
- A. J. Hall and P. Hodge, React. Funct. Polym., 1999, 41, 133 CrossRef CAS.
- P. Hodge and H. M. Colquhoun, Polym. Adv. Technol., 2005, 16, 84 CrossRef CAS.
- I. Goodman and B. F. Nesbitt, Polymer, 1960, 1, 384 CrossRef CAS.
- R. Nomura, A. Ueno and T. Endo, Macromolecules, 1994, 27, 620 CrossRef CAS.
- (a) M. F. Teasley, D. Q. Wu and R. L. Harlow, Macromolecules, 1998, 31, 2064 CrossRef CAS; (b) M. Chen and H. W. Gibson, Macromolecules, 1996, 29, 5502 CrossRef CAS.
- (a) D. Zhang, M. A. Hillmyer and W. B. Tolman, Macromolecules, 2004, 37, 8198 CrossRef CAS; (b) T. Fey, H. Keul and H. Höcker, Macromolecules, 2003, 36, 3882 CrossRef CAS; (c) D. Zhang, J. Xu, L. Alcazar-Roman, L. Greenman, C. J. Cramer, M. A. Hillmyer and W. B. Tolman, Macromolecules, 2004, 37, 5274 CrossRef CAS; (d) R. Slivniak and A. J. Domb, Biomacromolecules, 2005, 6, 1679 CrossRef CAS.
- (a) Y. F. Wang, K. P. Chan and A. S. Hay, Macromolecules, 1995, 28, 6371 CrossRef CAS; (b) M. G. Zolotukhin, S. Fomine, H. M. Colquhoun, Z. Zhu, M. G. B. Drew, R. H. Olley, R. A. Fairman and D. J. Williams, Macromolecules, 2004, 37, 2041 CrossRef CAS; (c) H. M. Colquhoun, M. G. Zolotukhin, Z. Zhu, P. Hodge and D. J. Williams, Macromol. Rapid Commun., 2004, 25, 808 CrossRef CAS.
- (a) Y. Hori, H. Hongo and T. Hagiwara, Macromolecules, 1999, 32, 3537 CrossRef CAS; (b) S. Okajima, R. Kondo, K. Toshima and S. Matsumura, Biomacromolecules, 2003, 4, 1514 CrossRef CAS.
- P. Hodge and S. D. Kamau, Angew. Chem., Int. Ed., 2003, 42, 2412 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1 CrossRef CAS.
- Z. Xue and M. F. Mayer, Soft Matter, 2009, 5, 4600 RSC.
- A. Gradillas and J. Pérez-Castells, Angew. Chem., Int. Ed., 2006, 45, 6086 CrossRef CAS.
- (a) G. Galli, G. Illuminati and L. Mandolini, J. Am. Chem. Soc., 1973, 95, 8374 CrossRef CAS; (b) G. Galli, G. Illuminati, L. Mandolini and P. Tamborra, J. Am. Chem. Soc., 1977, 99, 2591 CrossRef CAS; (c) L. Mandolini, J. Am. Chem. Soc., 1978, 100, 550 CrossRef CAS; (d) G. Ercolani, L. Mandolini, P. Mencarelli and S. Roelens, J. Am. Chem. Soc., 1993, 115, 3901 CrossRef CAS.
- S. Monfette and D. E. Fogg, Chem. Rev., 2009, 109, 3783 CrossRef CAS.
- J. E. Gautrot and X. X. Zhu, J. Mater. Chem., 2009, 19, 5705 RSC.
- S. D. Kamau, P. Hodge, A. J. Hall, S. Dad and A. Ben-Haida, Polymer, 2007, 48, 6808 CrossRef CAS.
- C. Y. Tastard, P. Hodge, A. Ben-Haida and M. Dobinson, React. Funct. Polym., 2006, 66, 93 CrossRef CAS.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 8424 CrossRef CAS.
- D. Xie, Q. Ji and H. W. Gibson, Macromolecules, 1997, 30, 4814 CrossRef CAS.
- (a) For example: D. J. Brunelle and T. G. Shannon, Macromolecules, 1991, 24, 3035 Search PubMed; (b) P. Hubbard, W. J. Brittain, W. J. SimonsickJr and C. W. Ross III, Macromolecules, 1996, 29, 8304 CrossRef CAS.
- C. L. Ruddick, P. Hodge, A. Cook and A. J. McRiner, J. Chem. Soc., Perkin Trans. 1, 2002, 629 RSC.
- A. Cook, P. Hodge, B. Manzini and C. L. Ruddick, Tetrahedron Lett., 2007, 48, 6496 CrossRef CAS.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760 CrossRef CAS.
- J. E. Gautrot and X. X. Zhu, Angew. Chem., Int. Ed., 2006, 45, 6872 CrossRef CAS.
- J. E. Gautrot and X. X. Zhu, Chem. Commun., 2008, 1674 RSC.
- Y. Yang and T. M. Swager, Macromolecules, 2007, 40, 7437 CrossRef CAS.
- S. Kang, B. M. Berkshire, Z. Xue, M. Gupta, C. Layode, P. A. May and M. F. Mayer, J. Am. Chem. Soc., 2008, 130, 15246 CrossRef CAS.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543 CrossRef CAS.
- E. W. Spanagel and W. H. Carothers, J. Am. Chem. Soc., 1935, 57, 929 CrossRef CAS.
- J. W. Hill and W. H. Carothers, J. Am. Chem. Soc., 1933, 55, 5031 CrossRef CAS.
- A. Ben-Haida, P. Hodge and H. M. Colquhoun, Macromolecules, 2005, 38, 722 CrossRef CAS.
- S. D. Kamau and P. Hodge, React. Funct. Polym., 2004, 60, 55 CrossRef CAS.
- H. M. Colquhoun, D. F. Lewis, P. Hodge, A. Ben-Haida, D. J. Williams and I. Baxter, Macromolecules, 2002, 35, 6875 CrossRef CAS.
- I. Baxter, A. Ben-Haida, H. M. Colquhoun, P. Hodge, F. H. Kohnke and D. J. Williams, Chem. Commun., 1998, 2213 RSC.
- A. Ben-Haida, H. M. Colquhoun, P. Hodge and D. J. Williams, Macromolecules, 2006, 39, 6467 CrossRef CAS.
- E. Thorn-Csanyi and K. Ruhland, Macromol. Chem. Phys., 1999, 200, 1662 CrossRef CAS.
- S. Sugihara, K. Toshima and S. Matsumura, Macromol. Rapid Commun., 2006, 27, 203 CrossRef CAS.
- H. Jakobson and W. H. Stockmeyer, J. Chem. Phys., 1950, 18, 1600 CrossRef CAS.
- Z. R. Chen, J. P. Claverie, R. H. Grubbs and J. A. Kornfield, Macromolecules, 1995, 28, 2147 CrossRef CAS.
- Handbook of Polyolefins, ed. C. Vasile, Marcel Dekker Inc., New York, 2nd edn, 2000, pp. 124–125 Search PubMed.
- Y. A. Ibrahim, J. Mol. Catal. A: Chem., 2006, 254, 43 CrossRef CAS.
- L. Reif and H. Höcker, Macromolecules, 1984, 17, 952 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS.
- M. R. Buchmeiser, Chem. Rev., 2009, 109, 303 CrossRef CAS.
- J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815 CrossRef CAS.
- D. E. Bergbreiter, J. Tian and C. Hongfa, Chem. Rev., 2009, 109, 530 CrossRef CAS.
- C. Hongfa, J. Tian, H. S. Bazzi and D. E. Bergbreiter, Org. Lett., 2007, 9, 3259 CrossRef CAS.
- I. K. Varma, A. C. Albertsson, R. Rajkhowa and R. K. Srivastava, Prog. Polym. Sci., 2005, 30, 949 CrossRef CAS.
- S. Matsumura, Adv. Polym. Sci., 2006, 194, 95 CAS.
- S. Kobayashi, Proc. Jpn. Acad., Ser. B, 2010, 86, 338 CrossRef CAS.
- A. Duda, A. Kowalski, S. Penczek, H. Uyama and S. Kobayashi, Macromolecules, 2002, 35, 4266 CrossRef CAS.
- L. van der Mee, F. Helmich, R. de Brujin, J. A. J. M. Vekemans, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2006, 39, 5021 CrossRef CAS.
- S. Namekawa, H. Uyama and S. Kobayashi, Proc. Jpn. Acad., Ser. B, 1998, 74, 65 CrossRef.
- B. J. B. Folmer, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2001, 123, 2093 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS.
- Y. F. Wang, K. P. Chan and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 375 CrossRef CAS.
- Q. Z. Guo, H. H. Wang, J. Y. Wu, D. Guo and T. L. Chen, Polym. Adv. Technol., 2010, 21, 290 CAS.
- H. M. Colquhoun, D. F. Lewis, R. A. Fairman, I. Baxter and D. J. Williams, J. Mater. Chem., 1997, 7, 1 RSC.
- Y. F. Wang, K. P. Chan and A. S. Hay, Macromolecules, 1996, 29, 3717 CrossRef CAS.
- Y. Z. Meng, S. C. Tjong and A. S. Hay, Polymer, 2001, 42, 5215 CrossRef CAS.
- H. M. Colquhoun, M. G. Zolotukhin, L. G. Sestiaa, F. Arico, Z. Zhu, P. Hodge, A. Ben-Haida and D. J. Williams, J. Mater. Chem., 2003, 13, 1504 RSC.
- K. M. Stridsberg, M. Ryner and A. C. Albertsson, Adv. Polym. Sci., 2002, 157, 41 CAS.
- S. D. Kamau, P. Hodge, R. T. Willimans, P. Stagnaro and L. Conzatti, J. Comb. Chem., 2008, 10, 644 CrossRef CAS.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927 CrossRef CAS.
- (a) M. J. Marsella, H. D. Maynard and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 1101 CrossRef CAS; (b) H. D. Maynard and R. H. Grubbs, Macromolecules, 1999, 32, 6917 CrossRef CAS.
- U. Lüning, F. Fahrenkrug and M. Hagen, Eur. J. Org. Chem., 2001, 2161 CrossRef CAS.
- P. A. Chase, M. Lutz, A. L. Spek, G. P. M. van Klink and G. van Koten, J. Mol. Catal. A: Chem., 2006, 254, 2 CrossRef CAS.
- W. Gao, R. Hagver, V. Shah, W. Xie, R. A. Gross, M. F. Ilker, C. Bell, K. A. Burke and E. B. Coughlin, Macromolecules, 2007, 40, 145 CrossRef CAS.
- I. N. A. Van Bogaert, K. Saerens, C. DeMuynck, D. Develter, W. Soetaert and E. J. Vandamme, Appl. Microbiol. Biotechnol., 2007, 76, 23 CrossRef CAS.
- S. K. Singh, A. P. Felse, A. Nunez, T. A. Foglia and R. A. Gross, J. Org. Chem., 2003, 68, 5466 CrossRef CAS.
- E. Zini, M. Gazzano, M. Scandola, S. R. Wallner and R. A. Gross, Macromolecules, 2008, 41, 7463 CrossRef CAS.
- J. E. Gautrot and X. X. Zhu, Macromolecules, 2009, 42, 7324 CrossRef CAS.
- H. Thérien-Aubin, J. E. Gautrot, Y. Shao, J. Zhang and X. X. Zhu, Polymer, 2010, 51, 22 CrossRef CAS.
- R. Mohr, K. Kratz, T. Weigel, M. Lucka-Gabor, M. Moneke and A. Lendlein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3540 CrossRef CAS.
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097 CrossRef CAS.
- R. A. Gross, B. Kalra and A. Kumar, Appl. Microbiol. Biotechnol., 2001, 55, 655 CrossRef CAS.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793 CrossRef CAS.
- H. Uyama, K. Takaya, N. Hoshi and S. Kobayashi, Macromolecules, 1995, 28, 7046 CrossRef CAS.
- (a) K. S. Bisht, L. A. Henderson, R. A. Gross, D. L. Kaplan and G. Swift, Macromolecules, 1997, 30, 2705 CrossRef CAS; (b) A. Kumar, B. Kalra, A. Dekhterman and R. A. Gross, Macromolecules, 2000, 33, 6303 CrossRef CAS.
- I. van der Meulen, M. de Geus, H. Antheunis, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2008, 9, 3404 CrossRef CAS.
- H. Uayama, K. Inada and S. Kobayashi, Polym. J., 2000, 32, 440 CrossRef.
- C. Berkane, G. Mezoul, T. Lalot, M. Brigodiot and E. Maréchal, Macromolecules, 1997, 30, 7729 CrossRef CAS.
- Y. Takahashi, S. Okajima, K. Toshima and S. Matsumura, Macromol. Biosci., 2004, 4, 346 CrossRef CAS.
- Y. Osanai, K. Toshima and S. Matsumura, Macromol. Biosci., 2004, 4, 936 CrossRef CAS.
- B. Manzini, P. Hodge and A. Ben-Haida, Polym. Chem., 2010, 1, 339 RSC.
- O. Noll and H. Ritter, Macromol. Rapid Commun., 1996, 17, 553 CAS.
| This journal is © The Royal Society of Chemistry 2011 |