Self-immolative linkers in polymeric delivery systems
Christopher A.
Blencowe
a,
Andrew T.
Russell
*a,
Francesca
Greco
b,
Wayne
Hayes
*a and
David W.
Thornthwaite
c
aDepartment of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, United Kingdom. E-mail: a.t.russell@reading.ac.uk; w.c.hayes@reading.ac.uk; Fax: +44 118 378 6331; Tel: +44 118 378 6234
bReading School of Pharmacy, University of Reading, Whiteknights, Reading, RG6 6AP, United Kingdom. E-mail: f.greco@reading.ac.uk; Fax: +44 118 378 4703; Tel: +0118 378 8244
cUnilever Research and Development, Quarry Road East, Bebington, Wirral CH63 3JW, United Kingdom
First published on 18th November 2010
Abstract
There has been significant interest in the methodologies of controlled release for a diverse range of applications spanning drug delivery, biological and chemical sensors, and diagnostics. The advancement in novel substrate-polymer coupling moieties has led to the discovery of self-immolative linkers. This new class of linker has gained popularity in recent years in polymeric release technology as a result of stable bond formation between protecting and leaving groups, which becomes labile upon activation, leading to the rapid disassembly of the parent polymer. This ability has prompted numerous studies into the design and development of self-immolative linkers and the kinetics surrounding their disassembly. This review details the main concepts that underpin self-immolative linker technologies that feature in polymeric or dendritic conjugate systems and outlines the chemistries of amplified self-immolative elimination.
 Christopher A. Blencowe | Christopher Blencowe obtained his Masters degree in Chemistry (1st class honours) from the University of Reading in 2003. During his Masters degree he undertook industrial positions at RSSL, GlaxoSmithKline and the University of Reading. His final year project involved the synthesis of self-immolative dendrimers for fragrance delivery applications. He is currently studying for a PhD under the guidance of Dr Wayne Hayes and Dr Andrew Russell developing novel self-immolative linker technologies. |
 Andrew T. Russell | Andy Russell received his undergraduate education at the University of Leicester, UK. He studied for his PhD under the guidance of Professor Garry Procter at the University of Salford before spending two years of post-doctoral studies in the USA. These were divided between Duke University and Case Western Reserve University supervised by Professors Richard Polniaszek and Anthony Pearson, respectively. Upon returning to the UK he became a Departmental Research Assistant in the Dyson Perrins Laboratory of the University of Oxford, working with Prof. Sir J. E. Baldwin FRS. His first academic appointment was at the University of Salford before moving to his present position at the University of Reading. His research program focuses on synthetic methodology and the total synthesis of natural products. |
 Francesca Greco | Francesca Greco obtained her Masters degree in Pharmacy (110/110 cum laude) from the University of Pisa in 2002. During her Masters, she carried out a one-year research project in industry at Pharmacia (now Pfizer), Milan, Italy. Then, she undertook her PhD project supervised by Prof. Duncan, Prof. Nicholson and Dr Vicent, at the Welsh School of Pharmacy, Cardiff University. In 2006, Francesca was appointed as Lecturer in Pharmaceutics at the School of Pharmacy, University of Reading. Her research on polymer-drug conjugates has led to publications, presentations at national and international conferences and prizes, including an invitation to the “Roche Symposium for leading scientists of the next decade” (Basel, Switzerland, 2007). |
 Wayne Hayes | Wayne Hayes is a graduate of Nottingham Trent University and the University of Birmingham where he carried out his PhD studies under the tutelage of Professor Sir J. Fraser Stoddart FRS. He currently holds the posts of Reader in Polymer Chemistry and Head of the Department of Chemistry at Reading. His research focuses on the utilization of supramolecular chemistry and modern polymerization methods to create novel polymeric materials such as self-assembling adhesives, healable networks, membranes for use in energy-related applications and hyperbranched drug delivery systems. In 2004 he was awarded the Young Researcher's Medal by MacroGroup UK for his work on novel dendritic and hyperbranched polymers. |
 David W. Thornthwaite | Dave Thornthwaite is a graduate of Sheffield University and completed his PhD there under the supervision of Dr J. McKenna on ‘enzyme model systems based on steroids’ in 1977. Since then he has worked for Unilever research for 33 years as an organic chemist covering areas of oxidation, surfactant, tea, peptide, perfume and catalytic chemistries as well as mechanistic work utilising kinetics of reaction and absorption onto substrates. He also provides advice on formulation stability and scale up of product manufacture. During this period he has supervised nine Unilever CASE sponsored PhD students and has taught industrial chemistry courses at City, Kent, Reading and Liverpool Universities as well as actively promoting young people into science careers. |
1. Introduction and key terminology
It has long been known that the reactivity or (biological) activity of an active compound (commonly a drug, referred to herein as a ‘reporter’ molecule) can be attenuated by the incorporation of a protecting group (also described as a ‘trigger’ group).1–4 Traditional protecting group strategies involve the direct connection of a protecting moiety with a compound via a scissile bond (Scheme 1a). This linkage is cleaved using specific conditions, for instance by enzymatic catalysis (e.g. peptidyl amides) or by ‘chemical modification’ (e.g. (bio)reduction of a nitro trigger), where the cleavage mechanism and conditions are determined by the nature of the protecting group. This classical prodrug approach has been widely explored for small molecules5,6 as well as macromolecular conjugate systems.7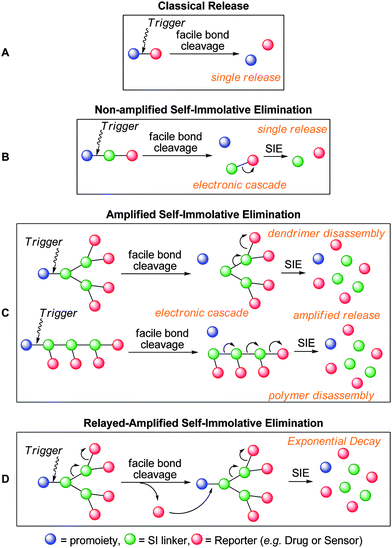 | ||
| Scheme 1 (a) (Bio)chemical cleavage of a two-component pro-moiety, b) self-immolative elimination of a three component pro-moiety, c) amplified disassembly of self-immolative polymeric promoieties, d) relayed amplified self-immolative elimination. | ||
This strategy works very well when the linkage is easily accessible. However, when the trigger and/or the reporter moieties are sterically bulky, their spatial proximity can impair cleavage, thus preventing release.8,9 To overcome this problem, an alternative strategy has been suggested whereby an additional linker is incorporated between the trigger and the reporter moiety. In this approach, the linker forms a scissile bond with a protecting group and a stable bond to a reporter group, the latter of which becomes labile upon removal of the protecting group, resulting in rapid disassembly of the three components (Scheme 1b). This linker technology is becoming increasingly common in conjugate systems for drug delivery and is referred to as ‘self immolative linkers’.10–13
Recent reviews have discussed self-immolative linkers for tumour-activated prodrug therapies and biological conjugates,14–17 herein we systematically review the mechanistic aspects of self immolative technology focussing on their use in macromolecular systems.
Classical self-immolative linkers combine a protecting group with a single reporter moiety, whereby a single activation event leads to the release of a single reporter group. This release can be described as non-amplified. The evolution of self-immolative linker technology has led to self immolative systems capable of amplified release (Scheme 1c). In this case a single activation event leads to the release of multiple reporter groups.18–20 More recently, systems capable of relayed amplified release whereby a single activation event facilitates exponential release have been developed (Scheme 1d). Examples of such system amplification are discussed in relevant sections.
For the purpose of clarity, a colour scheme has been adopted to highlight trigger (blue), self-immolative linker (green) and reporter (red) moieties.
2. Classes of self-immolative elimination
2.1. Self-immolative elimination
Self-immolative elimination is the spontaneous and irreversible disassembly of a multicomponent compound into its constituent fragments through a cascade of electronic elimination processes. Self-immolative elimination is driven by two processes:- i) an increase in entropy coupled with ii) the irreversible formation of thermodynamically stable products (e.g. CO2).This phenomenon is most commonly observed for polysubstituted, electron-rich aromatic species that feature an electron-donating substituent (such as amino or hydroxyl residues) that is in conjugation (ortho or para) with a suitable leaving group based at a benzylic position. An electron-donating substituent is required commonly to lower the energy barrier of dearomatisation. However, for compounds of this type, the incorporation of a protecting group or trigger is often required to modulate the spontaneity of self-immolative elimination under physiological or basic conditions. For example, Sauerbrei and co-workers have demonstrated the self-immolative ability of a p-hydroxylbenzyl alcohol based linker with the controlled release of a series of tetrahydro-β-carbolines from a solid-phase polymer conjugate (Scheme 2).21 In its ‘pro’ form, the phenolester of 1 is insufficiently electron-donating to facilitate self-immolative elimination. However, enzymatic cleavage of the scissile ester bond and enzyme assisted deprotonation of the corresponding phenol, afforded a strongly electron-donating phenoxide 2 that facilitates the formation of quinone methide intermediate 3 following an 1,6-elimination process. In the presence of solvent (e.g. H2O or MeOH), solvolytic quenching of 3 restored the aromaticity to form p-hydroxybenzyl methyl ether 4. The presence of a nucleophile or polar protic solvent is required to facilitate fragmentation and leads to concomitant quenching of the high energy, highly electrophilic intermediate. When the nucleophile is the solvent, self-immolative elimination is alternatively described as a process of solvolytic cleavage (or solvolysis). Polar protic solvents such as methanol are commonly employed in these studies and also serve as diagnostic probes in mechanistic degradation assays.
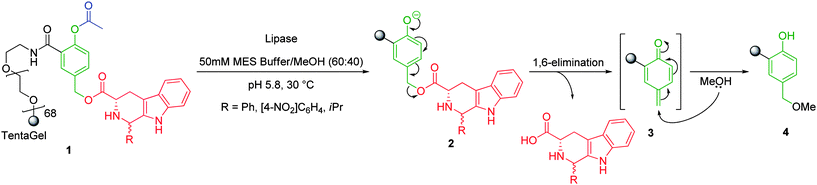 | ||
| Scheme 2 Lipase mediated self-immolative elimination of tetrahydro-β-carbolines from tentagel solid support 1.21 | ||
The elimination process may be described as a quinone (for self-immolative phenols) or an azaquinone (for self-immolative anilines) methide mediated elimination-addition reaction which describes the nature of the formally charged intermediate that is formed from disassembly.
A range of benzyl alcohols have been thoroughly investigated as a self-immolative linkers including 2- and 4- amino-,10 hydroxy-12,13 and mercapto-11,22 functionalised benzyl alcohols (based on 5–7, Fig. 1). The reduced self-immolative ability of 2- and 4-hydroxybenzyl alcohol based linkers, as a result of the reduced nucleophilicity of oxygen with respect to nitrogen, can be enhanced by the presence of additional activating ring substituents or mildly basic conditions. The superior self-immolative ability of the aminobenzyl alcohol linker has favoured its use, prompting detailed kinetic studies regarding ring and methylene substituted analogues.23–25 Hay and co-workers have reported that the rate of self-immolative elimination of the p-aminobenzyl alcohol linker is enhanced by the presence of electron-donating substituents or halides, and impaired by the presence of electron-withdrawing substituents. This effect is most strongly influenced when these substituents are in conjugation with the leaving group at a benzylic position and thus provide the greatest degree of (de)stabilisation of the azaquinone methide intermediate.
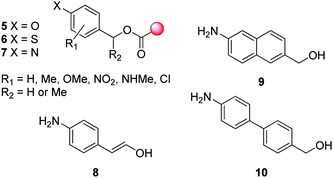
The most predominant enhancement in elimination rate was observed when additional substituents (e.g. methyl) were incorporated at the benzylic methylene position (the solvolytic site), which stabilises the charged intermediate species by hyperconjugation.
Despite the observation made by De Groot and co-workers of the elongated 1,8-self-immolative elimination of p-aminocinnamylalcohol 8,25 the distance over which self-immolative elimination can propagate has really restricted this phenomenon to monocyclic-aromatic systems. De Groot and co-workers have shown that naphthyl- 9 and biphenyl- 10 (bicyclic-aromatic) based self-immolative linkers are not susceptible to their corresponding 1,8- and 1,10-eliminations under aqueous conditions or even at elevated temperatures.26
Despite the structural and electronic limitations of the self-immolative elimination process, the development of self-immolative linkers has led to a diverse range of homoaromatic10,11,13,28 coumarin,29 furan,30 thiophene,30 thiazole,30 oxazole,30 isoxazole,30 pyrrole,30 pyridine,31 pyrazole,30 imidazole,32,33 and triazole34 based heteroaromatic linkers that are self-immolative under both aqueous and physiological conditions.
2.2. Cyclisation elimination
The term “self-immolative elimination” has also been used to describe cyclisation-elimination reactions that result in molecular fragmentation via activation of a latent nucleophile followed by displacement of a carbonyl linked reporter moiety.35–37 In a manner analogous to self-immolative elimination, cyclisation-elimination is driven by i) the formation of thermodynamically stable fragments, which is combined with ii) an increase in entropy. This process has been observed most commonly for 4-aminobutanoyl esters and ethylenediamines. Cyclisation-elimination follows a series of equilibria and proton transfers which is strongly influenced by the environmental pH.38,39 This overall process is comparatively slow as reflected by prolonged half lives (typically 2–24 h). In contrast, the cyclisation-elimination of 2-hydroxy40 or 2-aminomethylbenzamides (11 and 12), 2-hydroxyhydrocinnamic esters41 and 2-hydroxyphenyl acetyl ester (13) based self-immolative cyclisation linkers is enhanced through the incorporation of substituents (such as bulky aromatic or gem-dimethyl residues) which enforces a biased population of reactive conformations (Fig. 2).42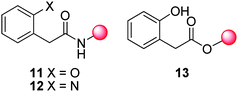 | ||
| Fig. 2 Amino- 11 or hydroxyl- 12 methylbenzamide and hydroxyl phenylacetyl ester 13 based self-immolative linkers.40 | ||
Self-immolative cyclisation-elimination linkers have proven advantageous for the release of amino- or hydroxyl-based compounds from hydrolytically stable amide or carbamate linkages. For example, Shabat and Amir have demonstrated the release of p-nitrophenol from a first generation diethylene triamine based dendron (Scheme 3).27 Amide 14 is highly stable under physiological conditions. However, the selection of the amide side chain of penicillin G affords an elegant solution to the need for mild cleavage of an otherwise stable functional group. Thus cleavage of either (or both) phenylacetamide protecting groups by penicillin-G-amidase enzymes affords amine 15. This nucleophilic species then participates in a favourable 5-exo-trig (1,5-) cyclisation to liberate p-nitrophenol (the reporter unit in this model study) with concomitant formation of imidazolidinone 16.
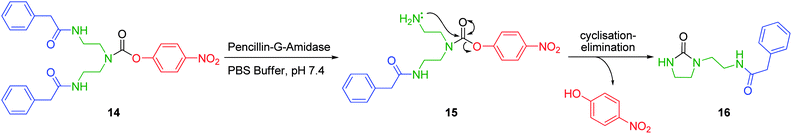 | ||
| Scheme 3 PGA mediated cyclisation elimination of 4-nitrophenol from first generation diethylenetriamine based dendron 14.27 | ||
2.3. Amplified self-immolative elimination
Amplified self-immolative elimination is defined by the release of multiple reporter groups via a series of electronic elimination processes upon a single activation event.18–20 The incorporation of additional leaving groups into a linear self-immolative linker creates a branched self-immolative linker of type ABn (where A represents an electron-donating group and B represents suitable leaving groups) capable of amplified release. For example, De Groot and co-workers have reported the self-immolative elimination of 2-phenylethanol from a first generation dendron featuring a 2,4,6-tris(hydroxymethyl)aniline AB3 branched linker (Scheme 4).18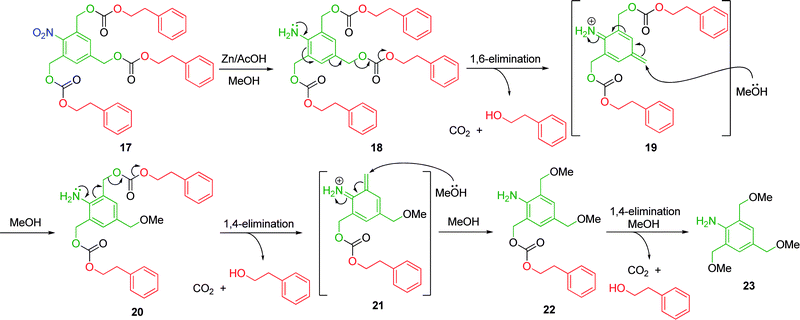 | ||
| Scheme 4 Triple self-immolative elimination of 2-phenylethanol from first generation 2,4,6-tris(hydroxymethyl)aniline based AB2 dendron 17.18 | ||
Dendron 17, when subjected to zinc in the presence of acetic acid, is reduced to afford aniline 18, which, by dint of its low pKa has a significant percentage in the non-protonated form and so, in turn undergoes a 1,6-elimination with concomitant decarboxylation to liberate 2-phenylethanol (the reporter molecule) and azaquinone methide 19. Quenching with solvent reforms the electron-donating aniline which undergoes two sequential 1,4-elimination and concomitant decarboxylation reactions to liberate two additional 2-phenylethanol reporter molecules. Overall, three 2-phenylethanol molecules were released from a single reduction reaction. To prove the concept of amplified self-immolative elimination the reaction was conducted in MeOH. The use of an organic polar protic solvent as opposed to an aqueous system has proven to be an effective method to determine that self-immolative elimination and solvent quenching of the azaquinone methide intermediate does occur preferentially with respect to direct cleavage of the carbonate linkage. In the former case, solvent quenching with methanol affords benzylic methyl ether 23, whilst direct cleavage of the carbamate linkage would yield the corresponding benzylic triol.
Only a few kinetic studies have been conducted upon branched self-immolative linkers, possibly as a result of the presumption that the differences in kinetic profiles between linear and branched linkers are insignificant. However, Erez and Shabat have conducted a systematic study on the relative rates of 1,4- and 1,6-elimination from 1,2- and 1,4- substituted linear- and 2,4-bis(hydroxymethyl)aniline AB2 branched linkers.43 The relative disassembly rates of phenylacetamide protected dendrons 24 and 25 (Fig. 3), which featured p-nitroaniline and 5-amino-2-nitrobenzoic acid in alternating positions highlighted a small difference between rates of 1,4- and 1,6-elimination. A step-wise kinetic profile was observed, whereby release of the ortho substituent was slowed by competitive 1,6-elimination until release of the para substituent had proceeded to completion. This is indicative of a step-wise mechanism which follows preferential solvolysis of the para substituent (see Scheme 4). A larger discrepancy (two fold difference between 1,4- and 1,6-eliminations) was observed for the relative elimination rates of 26 and 27, which cannot be attributed to their relative steric bulk acting to hinder enzymatic hydrolysis.
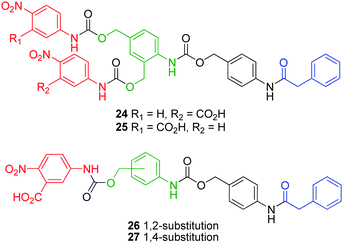 | ||
| Fig. 3 Linear aminobenzyl alcohol and AB2 branched 2,4-bis(hydroxymethyl)aniline based self-immolative systems.43 | ||
Instead, the accumulation of the ortho substituted aniline intermediate, observed from the degradation of 26, suggests that 1,4-elimination of the ortho substituent was the rate limiting process in this degradation study. Shabat and co-workers have also conducted a study regarding substituent effects upon the rates of the first order disassembly of 2,6-bis(hydroxymethyl)phenol based AB2 dendrons.44 It was shown that the incorporation of a para ethylester enhanced the rate of self-immolative elimination (2.88 ± 0.06 h−1) through electron-withdrawing resonance stabilisation of the anionic charge which develops on the corresponding phenol prior to quinone methide rearrangement. The relative disassembly rate of the methyl substituted phenol 29 (0.089 ± 0.002 h−1) was reduced approximately 30 fold, as a result of the destabilisation of the phenoxide intermediate via inductive electron donation (Fig. 4). The difference in elimination rates (ca. 1.5 fold) between 28 and 29 correlated well with the pKa values calculated for p-cresol and 4-hydroxybenzoic acid (10.26 and 8.34, respectively).
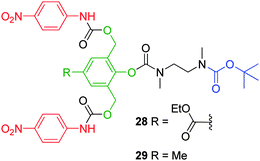 | ||
| Fig. 4 First generation 2,4-bis(hydroxymethyl)phenol AB2 based dendrons 28 and 29.44 | ||
3. Self-immolative polymeric systems
3.1. Self-immolative linear polymers
Whilst there has been substantial progress on the synthesis, development and assembly of polymers that are becoming increasingly similar to their natural and biologically significant counterparts, in terms of architecture, structure and function, minimal attention has been paid to methodologies of polymer disassembly under physiological conditions.45 This is surprising considering the ease with which natural polymers are assembled, modified and degraded. Thus, appropriately constructed polymers that are designed to undergo complete molecular disassembly upon a specific single activation event, to yield a high local concentration of an active species, would prove effective drug delivery vehicles and have already been shown to offer great promise as highly sensitive diagnostic tools.46,47Self-immolative polymers are constructed from the combination of multiple ABn self-immolative linkers in a linear fashion. A single activation event facilitates a linear cascade of the polymeric backbone which affords multiple active reporter species, directly (active monomers), or by sequential elimination from the side chain of each monomer. Based upon the development of linear self-immolative linkers and the unsurpassed self-immolative ability of aniline based linkers, suitably modified branched linkers based on these initial designs have been preferentially investigated and utilised. As demonstrated48 by Warnecke and Kratz self-immolative oligomers can be obtained via a itrative activation-coupling pathway. For example dimer 30, featuring a 2,4-bis(hydroxymethyl)aniline AB2 branched linker, was synthesised via selective protection of one B functionality as a t-butyldimethylsilyl ether, followed by the sequential activation by phenylchloroformate and coupling of an additional 2,4-bis(hydroxymethyl)aniline linker. Desilylation in the presence of HCl/MeOH followed by activation by phenylchloroformate and coupling of tryptamine afforded the desired dimer 30 in 7% overall yield via an eight step synthesis. Chemical reduction of the p-nitrobenzyloxycarbonyl trigger group, under neutral conditions, resulted in precipitation of the corresponding aniline 31, which prohibited comparative studies of the rate of disassembly with varying pH. However, dimer disassembly (via32) and liberation of tryptamine was observed under acidic conditions (Zn/AcOH (0.3%), H2O:MeCN (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), pH = 3), albeit at a slow rate, as a result of partial protonation of the active aniline species at this pH. This reduction in elimination rate facilitated an in-depth kinetic study of dimer 30 and related monomers 34 and 35, which highlighted three distinct steps to the fragmentation profile (Scheme 5). These were attributed to a rapid 1,6-depolymerisation to release the terminal tryptamine molecule prior to a phase of relative stagnation where little release was observed, followed by a constant but slower release of the side-chain tryptamine. This result was in agreement with previous studies43 which revealed that 1,6-elimination led to depolymerisation and that this occurs preferentially to 1,4-elimination (see Scheme 4 and Fig. 3). The stagnant phase was attributed to the build up and solvent quenching of the azaquinone methide intermediate to form active monomer 32 and facilitate the steady further release of tryptamine. First order kinetics with respect to dimer 30 were obtained which indicated an E1 type mechanism and the formation of a azaquinone methide intermediate.
:1), pH = 3), albeit at a slow rate, as a result of partial protonation of the active aniline species at this pH. This reduction in elimination rate facilitated an in-depth kinetic study of dimer 30 and related monomers 34 and 35, which highlighted three distinct steps to the fragmentation profile (Scheme 5). These were attributed to a rapid 1,6-depolymerisation to release the terminal tryptamine molecule prior to a phase of relative stagnation where little release was observed, followed by a constant but slower release of the side-chain tryptamine. This result was in agreement with previous studies43 which revealed that 1,6-elimination led to depolymerisation and that this occurs preferentially to 1,4-elimination (see Scheme 4 and Fig. 3). The stagnant phase was attributed to the build up and solvent quenching of the azaquinone methide intermediate to form active monomer 32 and facilitate the steady further release of tryptamine. First order kinetics with respect to dimer 30 were obtained which indicated an E1 type mechanism and the formation of a azaquinone methide intermediate.
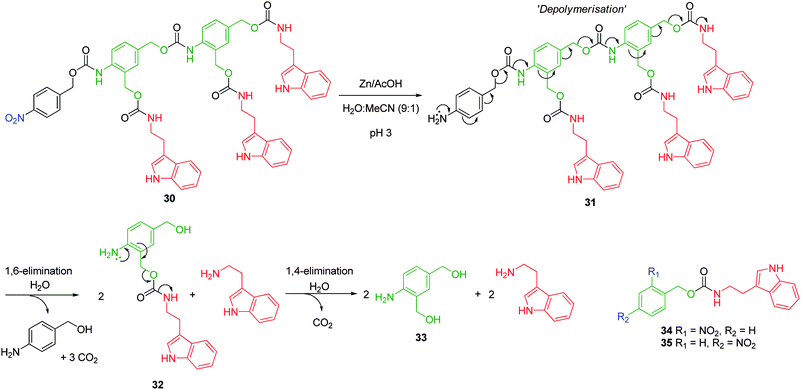 | ||
| Scheme 5 Self-immolative disassembly of 2,4-bis(hydroxymethyl)aniline based AB2 oligomer 30, inset – monomers 34 and 35.48 | ||
This difference between the rate of reaction for 1,6- and 1,4-elimination pathways was also observed by Shabat and co-workers in the two-stage disassembly of p-nitroaniline functionalised 3-(2-amino-5-(hydroxymethyl)phenyl)prop-2-en-1-ol based polymer 38 and copolymer 39 (Scheme 6), which is exemplified by the reduced elimination rate observed for the elongated, vinylogous side chain of 2-amino-5-hydroxymethylcinnamyl alcohol.49 Polymer 38, which featured an average degree of polymerisation (d.p.) of 11 (determined by NMR spectroscopic analysis), was synthesised from the homo-polymerisation of the corresponding phenyl carbamate (a blocked isocyanate),50 in the presence of dibutyltin dilaurate (DBTL) and was capped by the addition of 4-hydroxy-2-butanone. Polymer 38 was synthesised in 62% yield in one synthetic step. The repetitive connectivity of monomers through the p-amino substituent with the benzylic alcohol substituent via urethane linkages, created a polymer that was capable of complete disassembly through repetitive sequential 1,6-elimination and decarboxylation reactions. Polymer disassembly was demonstrated by piperidine mediated β-elimination of the 4-hydroxy-2-butanone protecting group in MeOH/DMSO. As also demonstrated with monomer 40, 1,6-elimination was comparably rapid (ca. 1 h) with respect to successive liberation of p-nitroaniline (ca. 20 h) via 1,6-elimination of the vinylogous side chain. The poor aqueous solubility of these polymers, as a result of their hydrophobic structure, prompted the synthesis of water-soluble copolymer 39.
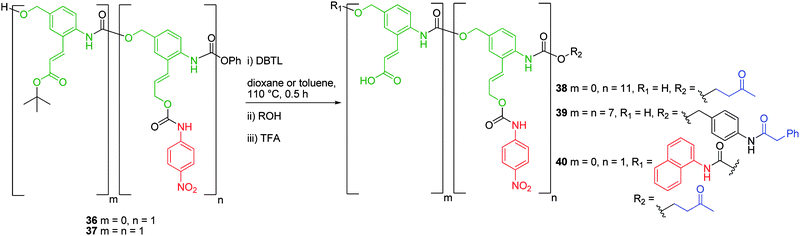 | ||
| Scheme 6 Synthesis of monomeric, oligomeric and polymeric 2-amino-5-hydroxymethylcinnamyl alcohol based AB2 polymers 38–40.49,51 | ||
Synthesised from the homopolymerisation of comonomer 37 in the presence of DBTL, copolymer 39 was prepared with an average degree of polymerisation of 7, and displayed enhanced solubility under physiological conditions. By preventing polymer aggregation, this facilitated the enhanced self-immolative elimination of p-nitroaniline (ca. 6 h) from its periphery upon incubation with a catalytic antibody.49
Linear self-immolative polymers of (2-amino-5-hydroxymethyl)cinnamic acid displayed both comparable aqueous solubility and rates of self-immolative elimination under physiological conditions.45,51 Weinstein and co-workers have demonstrated the potential of these polymers for enzyme detection. (2-Amino-5-hydroxymethyl)cinnamic acid exhibits a strong fluorescence which can be attenuated when the amino substituent is masked as an amide. In this study, oligomers 41 and 42 plus polymer such as 43, in the presence of an antibody, disassemble via an initial retro-Michael reaction and decarboxylation to liberate the free aniline intermediate 44. Subsequent depolymerisation affords a high local concentration of 45 which can be quenched by the nucleophilic addition of amino acid residues from the enzyme active site and surrounding surface to afford the aniline 46 (Scheme 7).52 This results in the formation of a covalent linkage between the enzyme and liberated fluorescent reporter molecule. The ability of 43 to label the enzyme without significant loss in activity was attributed to the capability of much of the polymer to diffuse away from the active site and react with distant nucleophilic residues on the enzyme surface, resulting in allosteric labelling, at low polymer concentrations (100 μM). Monomer 41 and trimer 42 also displayed limited labelling capabilities as a result of their ability to deactivate the enzyme via alkylation of the ε-amino lysine residue of the active site with minimal additional tagging.
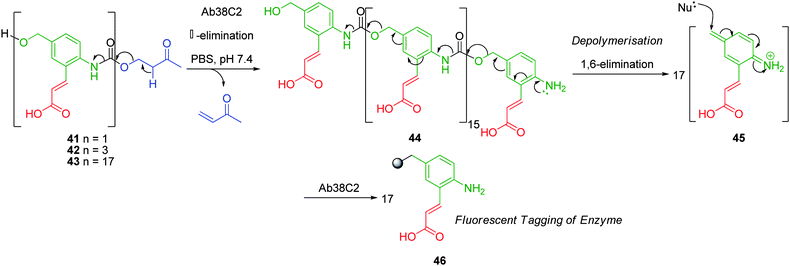 | ||
| Scheme 7 38C2 Antibody mediated self-immolative disassembly of linear (2-amino-5-hydroxymethyl)cinnamic acid based polymer 43.52 | ||
Esser-Kahn and co-workers have recently found that suitable side-chain modification of polymers based on the carbamate repeat unit (i.e.41) in conjunction with subsequent reaction with 2,4-toluene diisocyanate afforded cross-linked materials (Mn 15,600–17,300, PDI = 2.48–3.01) that were designed to rupture when exposed to appropriate chemical triggers.53
The sequential combination of multiple linear self-immolative linkers has also been demonstrated. In a seminal example reported by De Groot and co-workers, this approach has been employed as an effective method for maximising the enzymatic activity of prodrug conjugates through minimization of detrimental steric enzyme-prodrug interactions.26 More recently, DeWit and co-workers have developed methodologies for the condensation polymerisation of N,N′-dimethylethylenediamine, and p-hydroxybenzyl alcohol54 (d.p. 16, PDI = 1.58) or thioethanol55 (d.p. 35, PDI = 1.6), to afford linear self-immolative polymers capable of cascade depolymerisation upon removal of a single trigger moiety installed as a capping agent.
3.2. Self-immolative dendrimers
Dendrimers are perfectly branched and well-defined macromolecules which are characterised by high densities of terminal functional groups and compact, globular structures.56 These branched macromolecules are obtained from a step-wise synthetic approach whereby selective combination of ABn monomers via divergent57,58 or convergent59 growth provide macromolecules which possess very few structural defects and high degrees of molecular uniformity. The unique nature of the dendritic architecture and potential for high peripheral loading or encapsulation makes these branched macromolecules highly suitable candidates for applications including drug delivery, catalysis, sensors, imaging, gene therapy and solubilising agents.The study of the release of molecular species by covalent dissociation of dendrimers has led to the concept of ‘cleavable dendrimers’.60 Several groups have investigated the controlled self-immolative elimination of peripheral groups from the surface of dendrimers for fragrance delivery or sensing applications.61–63 Expansion of this concept has encompassed “self-immolative dendrimers”.18,20,64 Unlike conventional degradable dendrimers, where each molecule is released by an independent activation process, self-immolative dendrimers are able to release all peripheral molecular species (undergo complete fragmentation) in an amplified manner via a cascade of elimination reactions from a single activation event at the dendrimer core.
Self-immolative dendrimers are frequently synthesised via a divergent approach, which compliments the direction of the self-immolative cascade. Inclusion of a protecting group at the focal point facilitates the development of generations via a repetitive activation-coupling process. The size and thus generation attainable for self-immolative dendrimers is dependent upon the steric bulk of the reporter group and linker multiplicity (i.e. the ABn branched unit). Dendrimer solubility has also been found to be strongly influenced by the nature of the reporter group and linker hydrophobicity. Aniline based branched linkers have been utilised preferentially as a result of their unsurpassed self-immolative ability under physiological conditions and their ability to form carbamate linkages between the different generations. Phenol based linkers have also been employed as a consequence of their relative ease of synthesis, but require the incorporation of additional cyclisation elimination linkers such as ethylenediamine units to form stable carbamate linkages with trigger and reporter moieties.36
3.2.1.1. Aniline based AB2 linkers. De Groot and co-workers introduced the concept of branched self-immolative elimination with first and second generation dendrons featuring a 2-(4-aminobenzylidene)propane-1,3-diol AB2 self-immolative linker that was capable of degrading via two 1,8-elimination processes from a single activation event.18 The synthesis of dendron 47 (Fig. 5), starting from 2-(4-nitrobenzylidene)propane-1,3-diol, facilitated the divergent growth of multiple generations via alternate acyl transfer with p-nitrophenylchloroformate followed by coupling of additional branched linker. Paclitaxel (Taxol), was utilised as the model reporter group and attached via the intermediate p-nitrophenylcarbonate. Dendron 47 was obtained in 12% yield overall via a four step synthesis. The presence of an aniline based substituent resulted in the formation of stable carbamate linkages between dendron generations, and afforded a self-immolative dendron capable of disassembly under mildly acidic conditions (Zn/AcOH in MeOH). The validity of the self-immolative ability of 47 was examined by analogous reductive self-immolative elimination experiments conducted on 2′-O-(cinnamyloxycarbonyl) paclitaxel, which confirmed that the absence of an amino substituent inhibited drug release. The self-immolative ability of the 2-(4-aminobenzylidene)propane-1,3-diol branched linker was comparable with that of the linear p-aminocinnamyl alcohol linker capable of undergoing analogous 1,8-elimination in a manner similar to 38–40 (Scheme 6).25
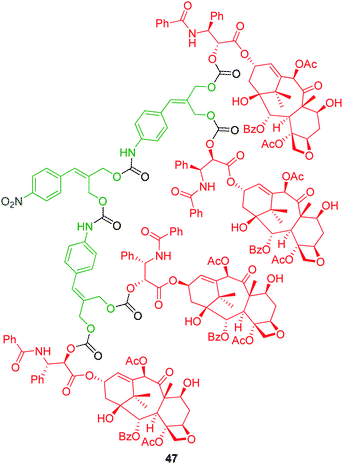 | ||
| Fig. 5 Second generation 2-(4-aminobenzylidene)propane-1,3-diol AB2 based dendron 47.18 | ||
3.2.1.2. Aniline based AB3 linkers. Shabat and co-workers have demonstrated the specific pencillin-G-amidase mediated hydrolysis of phenylacetamide protected, self-immolative, AB3 functionalised, first generation dendrons to release tryptophan and melphalan under physiological conditions (based on 48, Fig. 6).65 Furthermore, utilising the same 2,4,6-tris(hydroxymethyl)aniline AB3 branched linker capable of undergoing triple elimination, Shabat and Sella have demonstrated the heightened sensitivity of fluorogenic probes designed to detect triacetone triperoxide explosives.47
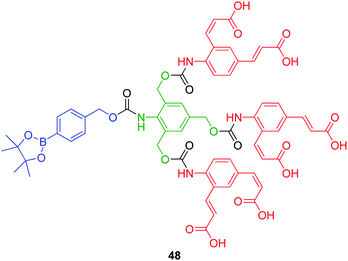 | ||
| Fig. 6 First generation 2,4,6-tris(hydroxymethyl)aniline AB3 based dendron 48.47 | ||
2,4,6-Tris(hydroxymethyl)aniline AB3 based dendron 48 (Fig. 6) featuring the use of a borate ester as a masked hydroxyl group, was shown to undergo complete disassembly upon oxidation of the C–B bond and treatment with alkaline hydrogen peroxide. As a result of its dendritic platform, amplified release of three equivalents of peripheral (4-amino-1,3-phenylene)diacrylic acid via a single 1,6- and then two 1,4-elimination processes, facilitated detection of triacetone triperoxide at the microgram level by fluorescence spectroscopic analysis and upon unmasking of the acetylated aniline. As expected, self-immolative dendron 48 was reported to be 3-fold more sensitive than its monomeric analogue as well as the corresponding borate ester functionalised self-immolative sensors featuring peripheral p-nitroaniline and 7-amino-4-methylcoumarin studied by Lo and Chu.66
3.2.1.3. Phenol AB2 based linkers. Szalai and co-workers have demonstrated that the self-immolative ability of the 2,4-bis(hydroxymethyl)phenol AB2 linker under mildly basic conditions is sufficient to mediate the disassembly of ether linked first and second generation 2,4-bishydroxymethyl)phenol based dendrons.20,67 An independent observation reported by Senter and co-workers highlighted that replacement of a carbonate linkage with an ether moiety impacts significantly on the rate of self-immolative elimination of the incorporated linker.68 This limits such linkers to the release of amino or hydroxyl substituted aromatic units, which have better abilities to act as leaving groups as a consequence of their reduced pKa values (cf. aliphatic amines and alcohols).68,69
Szalai and co-workers devised an efficient synthetic methodology, which was applied to the divergent synthesis of p-nitrophenol functionalised dendrons based on 49 (Fig. 7).20,67 By selective protection of dimethyl 4-hydroxyisophthalate or 2,4-diformylphenol, successive generations were compiled by repetitive reduction, chlorination and etherification. The self-immolative ability of dendron 49, obtained in 23% yield (over seven steps), was demonstrated by the release of p-nitrophenol via photolytic cleavage of the o-nitrobenzylether protecting group followed by disassembly of the corresponding phenol under mildly basic conditions. Degradation of dendron 49 was monitored by 1H NMR spectroscopic analysis, which revealed the formation of 2,4-dimethylphenol from quenching of the quinone methide intermediate with sodium borohydride in DMSO-d6.
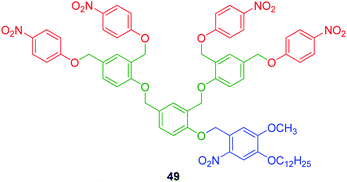 | ||
| Fig. 7 Second generation 2,4-bis(hydroxymethyl)phenol AB2 dendron 49.20,67 | ||
2,6-Bis(hydroxymethyl)-p-cresol is a commercially available AB2 branched linker capable of double 1,4-elimination and is a structural variant of the 2,4-bis(hydroxymethyl)phenol AB2 linker. Shabat and co-workers first demonstrated the self-immolative ability of this linker with the synthesis of first and second generation dendrons based on 50 (Fig. 8), which featured a photolabile protecting group joined through an ethylenediamine cyclisation linker and peripheral aminomethylpyrene.19 Photochemical cleavage of the protecting group initiated spontaneous cyclisation of the ethylenediamine spacer to afford 1,3-dimethylimidazolidin-2-one and the unprotected phenol, which under mildly basic conditions (10% triethylamine (TEA) in MeOH), underwent 1,4-elimination and decarboxylation to liberate an aminomethylpyrene molecule via quinone methide formation. Solvolytic quenching and reformation of the phenol, facilitated a second 1,4-elimination and concomitant decarboxylation to liberate a second aminomethylpyrene molecule and 2,6-bis(hydroxymethyl)-p-cresol. The slightly basic conditions were necessary to facilitate self-immolative elimination via the formation of the more reactive phenoxide following deprotection. The equivalent third generation dendron could not be synthesised as a result of the limited loading capacity of higher generation dendrimers for sterically bulky peripheral substituents as a consequence of the compact structure of the dendron surface. However, the corresponding p-nitroaniline functionalised third generation dendron featuring a t-butyloxycarbonyl trigger, separated by a ethylenediamine based cyclisation linker, was synthesised and was shown to undergo complete disassembly to release eight molecules of p-nitroaniline under analogous conditions upon a single trifluoroacetic acid (TFA) mediated cleavage of the t-butyloxycarbonyl group followed by neutralisation with TEA.
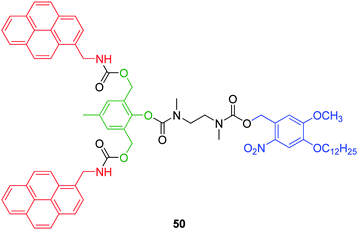 | ||
| Fig. 8 First generation 2,4-bis(hydroxymethyl)-4-cresol based AB2 dendron 50.19 | ||
Fomina and co-workers have utilised 2,6-Bis(hydroxymethyl)-p-cresol featuring a UV and near-IR sensitive ortho-nitrobenzylether trigger for the synthesis of polymeric nanoparticles.70 Copolymerisation with adipoyl chloride afforded polyesters (up to 65,000 Da, PDI = 1.54) capable of encapsulating Nile Red. Irradiation at 350 or 750 nm resulted in the self-immolative elimination of cresol units and the perforation of these nanoparticles.
3.2.1.4. Phenol AB3 based linkers. Extending the results from the 2,6-bis(hydroxymethyl)-p-cresol AB2 linker, 2,4,6-tris(hydroxymethyl)phenol is able to facilitate triple elimination of substituents via two 1,4-elimination processes and an additional 1,6-elimination pathway through the corresponding p-hydroxylmethyl group in a manner reminiscent of De Groot's 2,4,6-tris(hydroxymethyl)aniline 17 (see Scheme 4). Haba and co-workers have reported the synthesis and self-immolative elimination of a first generation 2,4,6-tris(hydroxymethyl)phenol based dendritic heterotrimeric prodrug (Scheme 8).71 Employing a divergent synthetic approach facilitated the step-wise coupling of doxorubicin directly through its primary amine and etoposide and camptothecin via the bisamine cyclisation linkers. To account for the steric bulkiness of the peripheral substituents, the enzyme labile trigger moiety was joined through a series of linear p-hydroxybenzyl alcohol and bisamine cyclisation linkers. The self-immolative elimination of dendron 51 was demonstrated under physiological conditions in the presence of catalytic antibody 38C2 enzymes, which facilitated dendron disassembly through retro-aldol, retro-Michael cleavage of the trigger group. In this seminal tritherapy study, three different drugs were released from a single molecular scaffold, via a 1,6- and two 1,4-elimination processes, using only a single activation event and this approach proved more effective than the individual monomeric or heterodimeric72 prodrugs when compared in vitro.71
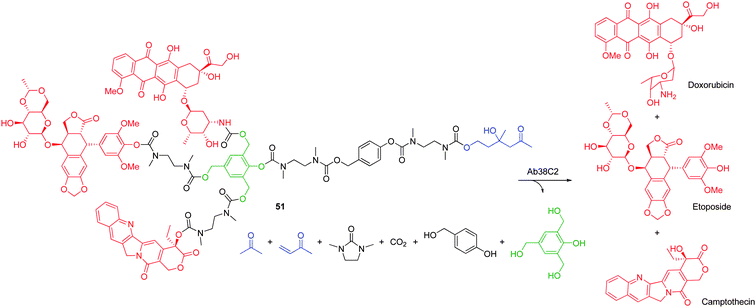 | ||
| Scheme 8 Triple self-immolative elimination of doxorubicin, etoposide and camptothecin from 2,4,6-tris(hydroxymethyl)phenol based AB3 dendron 42.71 | ||
3.2.1.5. Phenol AB6 based linkers. Solubility studies conducted on higher generation AB2 self-immolative dendrimers revealed that their high hydrophobicities tend to cause aggregation under aqueous conditions.73,74 This detrimental property poses a significant problem since the majority of systems developed to date have focussed around the area of controlled release for applications of biological importance. Shabat and Shamis have investigated the possibility of further increasing the multiplicity of branched linkers to afford lower generation self-immolative dendrimers with higher loading capabilities coupled with enhanced aqueous solubilities.74 Based upon 2-(4-aminobenzylidene)propane-1,3-diol, modification at the ortho positions afforded an AB6 branched linker (Fig. 9). Starting from 2,4,6-triformylphenol, dendron 52 was obtained via attachment of an N-t-butyloxycarbonyl (Boc) protected ethylenediamine trigger group, followed by sequential condensation with diethyl malonate and diisobutylaluminium hydride mediated reduction. Acyl transfer utilising p-nitrophenylchloroformate followed by displacement of p-nitrophenol with aminomethylpyrene afforded the desired hexa-functionalised first generation dendron in 1% yield overall. The self-immolative ability of this linker was demonstrated by the release of six molecules of pyrene (ca. 6 h), via four 1,6- and two 1,8-elimination processes mediated by TFA cleavage of the Boc protecting group, followed by neutralisation with tetrabutylammonium hydroxide.
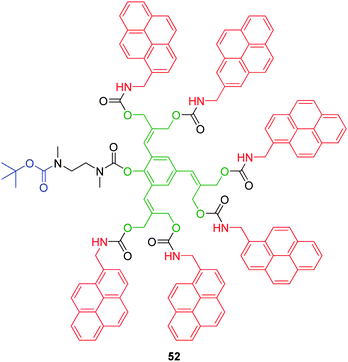 | ||
| Fig. 9 First generation AB6 dendron 52.74 | ||
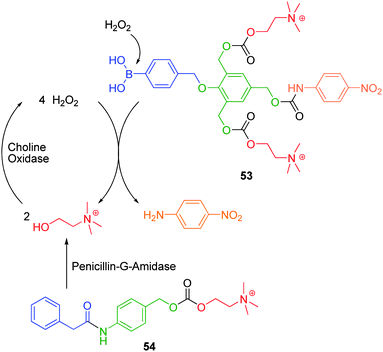 | ||
| Scheme 9 Relayed amplified self-immolative elimination of first generation 2,4,6-tris(hydroxymethyl)phenol based AB3 dendron 53.46 | ||
The liberation of choline and its subsequent oxidation to hydrogen peroxide by the addition of choline oxidase enzyme facilitated exponential self-immolative elimination of all of the dendrons present, at hydrogen peroxide concentrations as low as 5 μM, as determined by the UV spectroscopic analysis of liberated p-nitroaniline. Unfortunately, the sensitivity of this dendritic probe was limited by hydrolysis of carbonate bound choline in the absence of activating enzymes. Shabat and Sella have extended this concept of relayed amplified self-immolative elimination to detect the presence of activating enzymes. Incubation of 54 in the presence of penicillin-G-amidase enzymes led to the release of choline, which in the presence of 53 and choline oxidase, facilitated the exponential release of p-nitroaniline. Instead, the conjugation of peripheral methanol which could be oxidised to hydrogen peroxide by alcohol oxidase enzymes to facilitate exponential self-immolative elimination, afforded a conjugate system that proved relatively stable to hydrolysis and thus provided enhanced sensitivity.76 Sella and co-workers have extended this concept of relayed amplified self-immolative elimination to a two-component system, whereby the disassembly of the first component results in the exponential release of an analyte which facilitates the disassembly of a second component.77 Furthermore, the incorporation of a reducible quinone trigger moiety and peripheral mercaptoacetic acid has been shown to be an effective sensor for the detection of sulfhydryl compounds.78
Szalai and co-workers first demonstrated this concept with the disassembly of benzyl ether functionalised first and second generation dendrons that were capable of undergoing a linear electronic cascade upon allyl deprotection at the dendritic periphery to facilitate subsequent release of a single p-nitrophenol molecule from the dendron core.79 Whilst at first this principal seems counter-effective, Shabat and co-workers have reported that the electronic cascade can propagate from the periphery of one dendron, through the dendrimer focal point where it is amplified and facilitates the liberation of multiple reporter groups from the periphery of a second dendron.80,27
First and second generation dendrimers based on 55, have been shown to undergo complete disassembly under aqueous conditions upon a single triggering event at the dendron periphery. The suitably designed diethylenetriamine A2B branched cyclisation linker propagates a linear self-immolative cascade towards the dendrimer core, via the formation of an imidazolidinone, whilst a 2,6-bis(hydroxymethyl)phenol based AB2 branched linker facilitates amplified self-immolative elimination of peripheral 6-aminoquinoline (Scheme 10). The obvious advantage to this design is that multiple and different trigger moieties can be simultaneously incorporated to afford a self-immolative dendrimer that is sensitive to multiple orthogonally activating processes (e.g. different activating enzymes).75
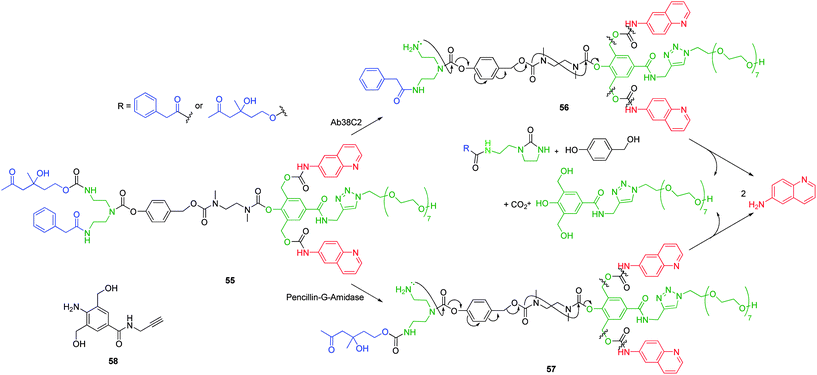 | ||
| Scheme 10 38C2 Antibody or PGA mediated self-immolative disassembly of 2,4-bis(hydroxymethyl)phenol based dendrimer 55, inset – monomer 58.75 | ||
3.3. Polymer-dendron conjugates
The tendency for higher generation self-immolative dendrimers to aggregate in aqueous solution has been an inherent problem with these systems and has prompted the development of strategies to improve dendrimer solubility. As a case in point, Shabat and co-workers have observed that enzymatic activation of higher generation dendrimers can be inefficient under physiological conditions.73,74 To negate this, simple modification of the leaving group (e.g. inclusion of ionizable side chains) can provide dramatic enhancements in dendrimer aqueous solubility.73Unfortunately, this is not a suitable strategy for the controlled release of bulky, hydrophobic reporter groups that are not amenable to structural modification. As a consequence, Shabat and co-workers have studied the release of highly hydrophobic chemotherapic agents from water soluble N-(2-hydroxypropyl)methacrylamide (HPMA) based conjugates that feature a 2,4,6-tris(hydroxymethyl)aniline, AB3 branched, linker that, in turn, is coupled through a Gly-Phe-Leu-Gly-Phe-Lys peptide and p-aminobenzyl alcohol self-immolative linker (Scheme 11).81 Synthesised in a linear fashion, starting from L-Boc-Phe-OH, the corresponding peptide protected AB3 prodrug was coupled to copolymeric HPMA (31,600 Da, PDI = 1.66) with an average loading of 7 taxol molecules per HPMA polymer (average of 21/3 dendrons) as determined by UV spectroscopic analysis (self-immolative dendritic loading capacity is typically ca. 4).18,19 The incorporation of a linear p-aminobenzyl alcohol self-immolative linker to maximise spatial separation of polymer and reporter moieties, was found necessary to relay enzymatic activity. As a consequence of its high loading self-immolative dendritic structure, polymer conjugate 59 exhibited superior antitumour activity with respect to traditional monomeric polymer-drug conjugates. To our knowledge this is the only example, to date, of a comb polymer conjugate which bears a self-immolative dendritic moiety that is capable of amplified release.
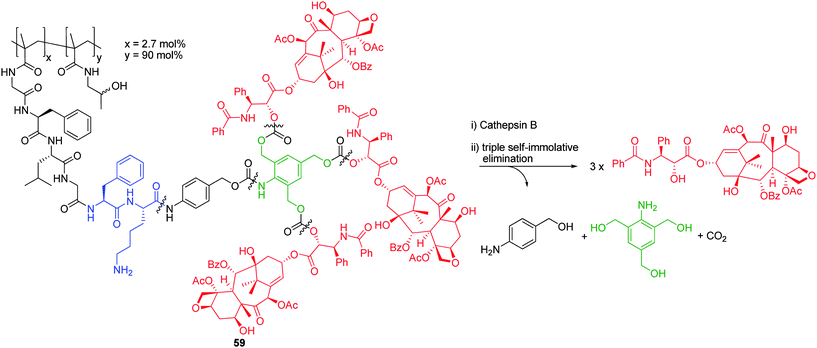 | ||
| Scheme 11 Cathepsin B mediated self-immolative disassembly of HPMA – 2,4,6-tris(hydroxymethyl)aniline based AB3 conjugate 59.81 | ||
Alternative studies have focussed on the modification of self-immolative dendrimers with solubilising polymers based on 55 (Scheme 10). Modification of the branched linker to incorporate solubilising polymers was considered as a suitable point of conjugation, since linker multiplicity and self-immolative ability would not be compromised. Amidation of 4-hydroxybenzoic acid with propargylamine followed by formylation in the presence of formaldehyde and sodium hydroxide afforded 4-hydroxy-3,5-bis(hydroxymethyl)-N-propargylbenzamide AB2 linker 58 which facilitated the coupling of an azido functionalised polyethyleneglycol (PEG) chain via an efficient copper-catalysed cycloaddition process. Formation of the corresponding triazole, mediated by copper (II) sulfate and copper (0) in the presence of tris((1-benzyl-triazol-4-yl)methyl)amine in DMF, afforded a highly water soluble polymer-dendron conjugate.82 Further work highlighted that the increased hydrophobicity of camptothecin functionalised dendrons, required extended PEG chains (up to 5000 Da) to provide adequate aqueous solubility and prevent aggregation in vitro.
The divergent synthetic approach undertaken with these conjugates facilitated the formation of 4-hydroxy-3,5-bis(hydroxymethyl)-N-propargylbenzamide based polymer-dendron conjugates bearing solubilising PEG (based on 55, Scheme 10) and dual UV-vis and fluorescence sensing abilities at the periphery.83 This route could enable the facile incorporation of alternative chromogenic reporter groups and thereby the type of signal output desired in the molecular probe. Furthermore, the nature of the trigger could be easily modified to allow for the highly sensitive detection of alternative enzymatic activities or chemical processes.
4. Non-amplified self-immolative polymeric conjugates
4.1 Linear self-immolative linkers in polymer conjugates
Self-immolative polymer conjugates may be defined as polymer conjugates which feature one or more non-amplified self-immolative linkers. The majority of research regarding self-immolative polymer conjugates has focussed on applications of biological relevance (e.g. drug delivery) and stems from pioneering studies on HPMA based conjugates conducted by Duncan and co-workers.84 Synthetic polymers (e.g. HPMA and PEG) have been widely used in a biological targeting role because of their well-established biocompatibility, non-immunogenicity, non-toxicity and excellent aqueous solubility.85,86 These polymers possess the added advantage of the versatility of synthetic chemistry that allows for molecular weight control and addition of biomimetic functions, in addition to the ability of passive tumour targetting via the enhanced permeability and retention effect.87 Self-immolative linkers had been combined with these polymers to afford self-immolative copolymeric conjugates.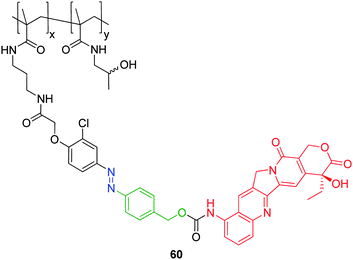
Greenwald and co-workers have conducted systematic studies on how the structures of amino- and hydroxy- substituted benzylalcohol based self-immolative linkers, and the nature of their conjugation with PEG prodrug conjugates of daunorubicin, effect the rate of enzyme mediated cleavage.91,92 The conjugation of PEG to p-aminobenzyl alcohol self-immolative linkers via hydrolytically stable amide or carbamate bonds (Fig. 12 – 63, 64) resulted in slow rates of hydrolysis. Carbamate linked conjugate 65, featuring p-hydroxybenzyl alcohol, displayed comparable hydrolytic lability with respect to carbonate 66 as these linkages have reduced resonance into the carbonyl from the phenol oxygen. As expected, ester bound conjugates 67–69 were most rapidly hydrolysed and as a consequence, conveyed the highest activity in vitro. The incorporation of methyl or methoxy substituents at the meta positions of the p-hydroxybenzyl alcohol self-immolative linker resulted in reduced drug activity, in both cases, as a result of these substituents exerting a steric effect at the site of enzyme hydrolysis. This contributed to a meta destabilising effect of the methoxy substituents on the benzylic position (σm = +0.12), whilst negating the stabilising effect of meta methyl substituents (σm = −0.07).
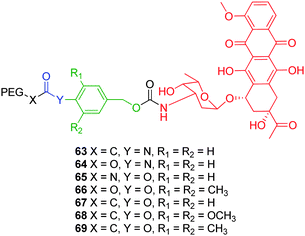 | ||
| Fig. 12 Self-immolative PEG-doxorubicin conjugates 63–69.91 | ||
Further studies by Greenwald and co-workers have focussed on the structure–activity relationships of PEG-daunorubicin conjugates featuring o-hydroxyhydrocinnamic acid derived cyclisation linkers.93 The incorporation of ring or β-substituents (e.g. gem-dimethyl) to this phenolic self-immolative linker has proven effective for increasing the population of the reactive conformer to facilitate 1,6-cyclisation.41,42 From the range of conjugates studied, in vivo evaluation highlighted that the arrangement of substituents about the self-immolative linker provided the most noticable enhancements in conjugate stability and drug efficacy. However, these enhancements were induced through steric interactions that prohibited enzymatic activation as opposed to facilitating enhancements in the cyclisation elimination process. Minimal variation of conjugate stability was observed from structural modification of the PEG chain or the nature of the polymer-self-immolative linker bond. Lee and co-workers have further applied these linker technologies to the selective self-immolative elimination of lysozyme enzymes from multi-PEGylated conjugates.94
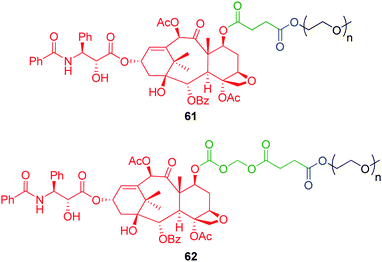
Branched polymer conjugates have also been developed which feature a bicin cyclisation-elimination linker.95,96 This A2B diethanolamine based branched linker facilitates the conjugation of two PEG polymer chains to a single reporter group which, in turn, has been shown to impart enhanced drug solubility and prolonged circulation times in vivo. PEG- drug and protein conjugates based on 70 have been shown to fragment via formation of morpholinolactones 71 or 72 upon a single enzyme activation (Scheme 12). In line with the results reported earlier by Suggs,97 following hydrolytic cleavage of the second PEG chain to form diol 73, an enhanced rate of cyclisation-elimination to afford 72, was observed. The reasons behind this rate enhancement are not completely clear. Reaction through the conformation of 73 proposed by Suggs allows approach of the alcohol on a Bürgi-Dunitz trajectory in a 6-exo-trig mode. In this conformation, with the second hydroxyethyl group equatorial, that hydroxyl group cannot reach the amide and its participation in the reaction appears limited to hydrogen bonding to the amine of the bicin linker. If this hydroxyethyl group is axial then an interaction with the amide carbonyl, through hydrogen bonding, is possible. Suggs's observation of reduction in hydrolysis rate, for the corresponding compound with a single hydroxyethyl group, by more than the statistical amount, suggests that both groups act cooperatively to enhance amide hydrolysis. To simplify the pharmacokinetic parameters, Filpula and co-workers modified the bicin linker to incorporate one PEG chain and an acetyl group, the latter being rapidly hydrolysed in vivo.96
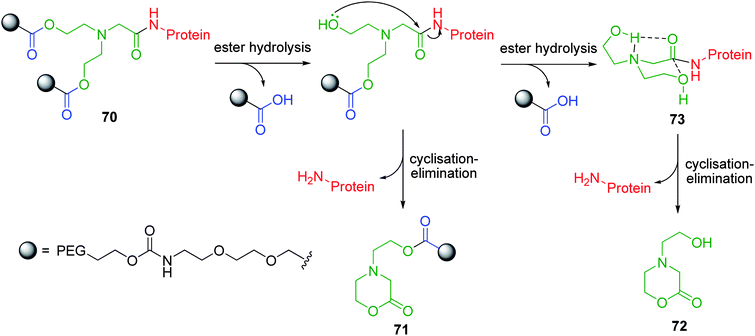 | ||
| Scheme 12 Cyclisation elimination of bicin based A2B PEG-protein conjugate 70.95,96 | ||
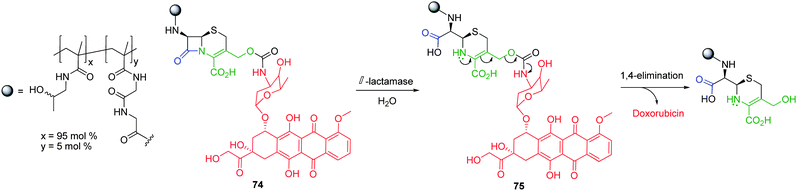 | ||
| Scheme 13 β-Lactamase mediated self-immolative elimination of cephalosporin based HPMA polymer conjugate 74.98 | ||
The cephalosporin self-immolative linkers are a rare example of a linker type which is connected to the polymeric support through a stable bond that is not cleaved concomitantly with activation. Shabat and co-workers have further developed this concept and have reported that simply modified linear self-immolative linkers can facilitate polymer conjugation via a chemically stable bond without compromising the reporter release (Fig. 13).100 Described as ‘chemical adapter systems’, these modified linkers commonly possess three different functionalities, the first acting as a handle to which a targeting or anchoring moiety is attached (e.g. polymer). The second functionality acts as a (bio)chemical trigger which facilitates the self-immolative elimination of a third functionality resulting in the liberation of a reporter group. In an initial study, Shabat and co-workers demonstrated the self-immolative ability of a HPMA based polymer conjugate of etoposide featuring 4-hydroxymandelic acid, a commercially available trifunctional linear self-immolative linker.101 Starting from 4-hydroxymandelic acid, conjugate 76 (Fig. 13) was synthesised in 13% overall yield over 6 steps by incorporation of a Boc protected-N,N′-dimethyldiethylenediamine side chain, followed by the addition of a protecting group via the corresponding p-nitrophenylcarbonate intermediate, capable of retro-aldol, retro-Michael elimination. Etoposide was incorporated through a ethylenediamine based linker, whilst TFA mediated deprotection of the side chain facilitated conjugation to polymeric HPMA.
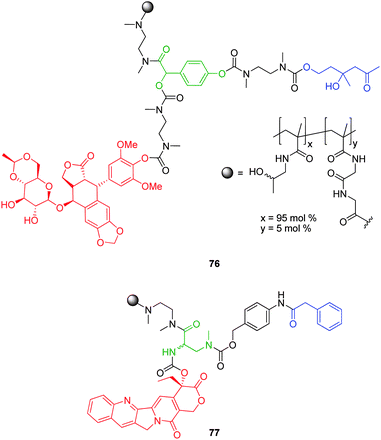 | ||
| Fig. 13 Self-immolative 4-hydroxymandelic acid and 2-amino-3-methylamino propanoic acid based HPMA polymer conjugates 76 and 77.101,102 | ||
Self-immolative elimination of etoposide from conjugate 76 was demonstrated under physiological conditions in the presence of antibody 38C2. Shabat and co-workers have also demonstrated the self-immolative ability of HPMA based conjugate 67 (Fig. 11) featuring a 2-amino-3-methylamino propionic acid linker, capable of cyclisation-elimination upon enzyme mediated activation.102 In both cases, UV spectroscopic analysis revealed low drug loadings (average of 3 per HPMA conjugate (30,000 Da)).101,102 This was attributed to an inefficient conjugation strategy which employed mild coupling conditions. However, it can be envisaged that the steric bulk of these chemical adaptor systems could hinder high loadings through congestion at the polymer surface.
The fact that these self-immolative linkers can facilitate the release of a reporter group but are themselves bound to a polymeric support that is separable, makes them highly suitable for solid-phase synthesis applications. Zheng and co-workers have investigated the solid-phase synthesis of short-chain peptides utilising a styrenic polymer conjugate which featured a reducible quinone capable of conformationally enhanced lactonisation cyclisation-elimination.103 Conjugate 78 facilitated the synthesis of C-terminal modified tetra- and penta-peptides in high yields (70–90%), which were isolated via mild chemical reduction (e.g. sodium hydrosulfite) of the quinone moiety followed by 1,6-cyclisation-elimination (Scheme 14). Conjugate 79 featured a chain extended tether located between the quinone and peptide linker moieties as utilised in the synthesis of N-terminal modified peptides.104 Zheng and co-workers have also shown that this self-immolative linker is highly stable to strong acidic and peptide coupling conditions, however, it undergoes facile cyclisation following mild chemical reduction (NaBH4) and tetrabutylammoniumfluoride mediated BAl2 type ester cleavage.
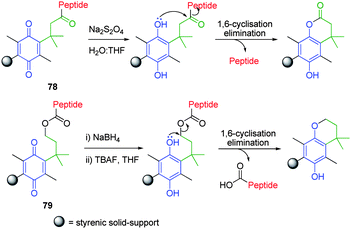 | ||
| Scheme 14 Reductive cyclisation elimination of oligomeric peptides from dihydroquinone based styrenic solid supports 78 and 79.103,104 | ||
Biocatalysed linkers have opened up advantageous alternatives to classical chemical approaches since enzymatic cleavage often occurs under very mild conditions and with pronounced selectivity.105 Waldmann et al. have studied the solid-phase synthesis and self-immolative abilities of polymer conjugates featuring a 2-(2-(aminomethyl)-4,5-dimethoxyphenyl)acetyl106 based cyclisation linker modified through the para ether substituent.107,108 The utility of conjugate 80 was demonstrated with the synthesis of a series of hydroxyl and amino based compounds utilising carbon-carbon cross coupling, Mitsunobu esterification and Diels–Alder cycloaddition processes. High isolated yields (60–94%) were obtained through facile pencillin-G-acylase mediated hydrolysis of the phenylacetamide protecting group, followed by cyclisation elimination of the reporter moiety from stable amide or ester bonds (Scheme 15).
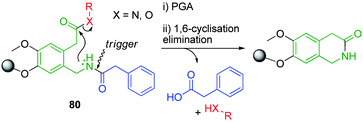 | ||
| Scheme 15 PGA mediated cyclisation-elimination of 2-(2-(aminomethyl)-4,5-dimethoxyphenyl)acetyl based solid support 80.107,108 | ||
Concluding remarks
In this article we have reviewed a structurally diverse range of self-immolative linkers which undergo cyclisation or electronic cascade reactions driven by entropic and thermodynamic factors. (See Table 1 for a summary of references categorised by self-immolative linker type). These linkers form stable bonds between a macromolecular scaffold and its reporter groups and are able to facilitate enhanced conjugate disassembly upon a specific activation event. The ability of these linkers to spontaneously fragment, yielding a high local concentration of active monomeric species, has been effectively exploited with amplified self-immolative polymer and dendrimer systems. Key studies in this area have highlighted the high loading capabilities of self-immolative dendrimers and the excellent solubility and stability characteristics of self-immolative polymers and self-immolative polymer conjugates. The blending of innovative strategies for release with skillful application of synthetic methods has driven the field forward. The ease with which these linkers can be modified and incorporated into macromolecular conjugates has made them applicable to a wide range applications spanning drug delivery, sensors and diagnostics. The development of relayed amplified self-immolative elimination, high multiplicity (e.g. AB6) branched linkers and efficient homopolymerisation processes has raised new opportunities for improved controlled delivery systems and exciting avenues for further investigation.| Size | Polymer Type | Linker Multiplicity | Mechanism of Disassembly | Signal Amplification | References |
|---|---|---|---|---|---|
| a Self-immolative elimination. b Cyclisation elimination. | |||||
| Small Molecules | N/A | AB | SIEa | Non-amplified | 9–13,23–26,28–34,66,68 |
| AB | CEb | Non-amplified | 35,38–42 | ||
| AB | SIE and CE | Non-amplified | 36,37 | ||
| Polymeric systems | Linear | AB | SIE | Non-amplified | 26 |
| AB | CE | Non-amplified | 55 | ||
| AB | SIE and CE | Non-amplified | 54 | ||
| AB2 | SIE | Amplified | 45,48,49,51–53,57 | ||
| Dendrimers | AB | CE | Non-amplified | 61–63 | |
| AB and AB2 | SIE | Non-amplified and Amplified | 43 | ||
| AB2 | SIE | Amplified | 18,20,67,73 | ||
| AB2 | SIE and CE | Amplified | 19,44,72 | ||
| AB2 | SIE | Relayed amplified | 46,76–78 | ||
| AB3 | SIE | Amplified | 47,65 | ||
| AB3 | SIE and CE | Amplified | 71 | ||
| AB6 | SIE | Amplified | 74 | ||
| AB | Receiver amplified | 79 | |||
| A2B | CE | Receiver amplified | 27,75 | ||
| (Mixed polymer types) | Polymer-dendron conjugates | AB3 | SIE | Amplified | 81 |
| A2B and AB2 | SIE and CE | Receiver amplified | 80 | ||
| Polymer conjugates | AB | SIE | Non-amplified | 21,22,88,89,91,92,94,98,99,101 | |
| AB | CE | Non-amplified | 90,93,102–104,106–108 | ||
| AB2 | SIE and CE | Amplified | 82,83,70 | ||
| A2B | CE | Receiver amplified | 95,96 | ||
| Antibody conjugates | AB | SIE | Non-amplified | 69 |
Acknowledgements
We thank EPSRC, Unilever and the University of Reading for a postgraduate studentship for CAB.Notes and references
- A. Albert, Nature, 1958, 182, 421 CrossRef CAS.
- N. J. Harper, J. Med. Chem., 1959, 1, 467 CrossRef CAS.
- A. K. Sinhababu and D. R. Thakker, Adv. Drug Delivery Rev., 1996, 19, 241 CrossRef CAS.
- V. J. Stella and K. J. Himmelstein, J. Med. Chem., 1980, 23, 1275 CAS.
- M. Skwarczynski, Y. Hayashi and Y. Kiso, J. Med. Chem., 2006, 49, 7253 CrossRef CAS.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688 CrossRef CAS.
- P. K. Chakravarty, P. L. Carl, M. J. Weber and J. A. Katzenellenbogen, J. Med. Chem., 1983, 26, 638 CrossRef CAS.
- F. M. H. De Groot, A. C. W. de Bart, J. H. Verheijen and H. W. Scheeren, J. Med. Chem., 1999, 42, 5277 CrossRef CAS.
- P. L. Carl, P. K. Chakravarty and J. A. Katzenellenbogen, J. Med. Chem., 1981, 24, 479 CrossRef CAS.
- P. D. Senter, W. E. Pearce and R. S. Greenfield, J. Org. Chem., 1990, 55, 2975 CrossRef CAS.
- G. Le Corre, E. Guibe-Jampel and M. Wakselman, Tetrahedron, 1978, 34, 3105 CrossRef CAS.
- L. D. Taylor, J. M. Grasshoff and M. Pluhar, J. Org. Chem., 1978, 43, 1197 CrossRef CAS.
- I. Tranoy-Opalinski, A. Fernandes, M. Thomas, J.-P. Gesson and S. Papot, Anti-Cancer Agent Med. Chem., 2008, 8, 618 Search PubMed.
- S. Papot, I. Tranoy-Opalinski, F. Tillequin, J.-C. Florent and J.-P. Gesson, Curr. Med. Chem.: Anti-Cancer Agents, 2002, 2, 155 CrossRef CAS.
- M. Avital-Shmilovici and D. Shabat, Soft Matter, 2010, 6, 1073 RSC.
- F. Kratz, I. A. Muller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20 CrossRef CAS.
- F. M. H. De Groot, C. Albrecht, R. Koekkoek, P. H. Beusker and H. W. Scheeren, Angew. Chem., Int. Ed., 2003, 42, 4490 CrossRef CAS.
- D. Shabat, R. J. Amir, N. Pessah and M. Shamis, Angew. Chem., Int. Ed., 2003, 42, 4494 CrossRef CAS.
- M. L. Szalai, R. M. Kevwitch and D. V. McGrath, J. Am. Chem. Soc., 2003, 125, 15688 CrossRef CAS.
- B. Sauerbrei, V. Jungmann and H. Waldmann, Angew. Chem., Int. Ed., 1998, 37, 1143 CrossRef CAS.
- S. Zalipsky, M. Qazen, J. A. Walker II, N. Mullah, P. Quinn and S. K. Huang, Bioconjugate Chem., 1999, 10, 703 CrossRef CAS.
- M. P. Hay, B. M. Sykes, W. A. Denny and C. J. O'Connor, J. Chem. Soc., Perkin Trans. 1, 1999, 2759 RSC.
- M. P. Hay, B. M. Sykes, D. Bohinc-Herceg, N. A. Helsby, C. J. O'Connor and W. A. Denny, J. Chem. Soc., Perkin Trans. 1, 2000, 1601 RSC.
- F. M. H. De Groot, E. W. P. Damen, T. J. Nevalainen, T. J. M. van den Bergh and H. W. Scheeren, Bioorg. Med. Chem., 2002, 10, 71 CrossRef.
- F. M. H. de Groot, W. J. Loos, R. Koekkoek, L. W. A. van Berkom, G. F. Busscher, A. E. Seelen, C. Albrecht, P. de Bruijn and H. W. Scheeren, J. Org. Chem., 2001, 66, 8815 CrossRef CAS.
- D. Shabat and R. J. Amir, Chem. Commun., 2004, 1614 RSC.
- S. Andrianomenjanahary, X. Dong, J.-C. Florent, G. Gaudel, J.-P. Gesson, J.-C. Jacquesy, M. Koch, S. Michel, M. Mondon and C. Monneret, Bioorg. Med. Chem. Lett., 1992, 2, 1093 CrossRef CAS.
- R. Weinstein, E. Segal, R. Satchi-Fainaro and D. Shabat, Chem. Commun., 2010, 46, 553 RSC.
- M. P. Hay, R. F. Anderson, D. M. Ferry, W. R. Wilson and W. A. Denny, J. Med. Chem., 2003, 46, 5533 CrossRef CAS.
- R. Perry-Feigenbaum, P. S. Baran and D. Shabat, Org. Biomol. Chem., 2009, 7, 4825 RSC.
- M. A. Naylor, S. A. Everett, K. B. Patel, M. R. L. Stratford and P. Wardman, Bioorg. Med. Chem. Lett., 1999, 1267.
- M. P. Hay and W. A. Denny, Tetrahedron Lett., 1997, 38, 8425 CrossRef CAS.
- P. Bertrand and J. P. Gesson, J. Org. Chem., 2007, 72, 3596 CrossRef CAS.
- D. Shabat, H. N. Lode, U. Pertl, R. A. Reisfeld, C. Rader, R. A. Lerner and C. F. Barbas III, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7528 CrossRef CAS.
- F. M. H. De Groot, L. W. A. van Berkom and H. W. Scheeren, J. Med. Chem., 2000, 43, 3093 CrossRef CAS.
- D. Shabat, N. Pessah, M. Reznik, M. Shamis, F. Yantiri, H. Xin, K. Bowdish, N. Shomron and G. Ast, Bioorg. Med. Chem., 2004, 12, 1859 CrossRef CAS.
- W. S. Saari, J. E. Schwering, P. A. Lyle, S. J. Smith and E. L. Engelhardt, J. Med. Chem., 1990, 33, 97 CrossRef CAS.
- W. S. Saari, J. E. Schwering, P. A. Lyle, S. J. Smith and E. L. Engelhardt, J. Med. Chem., 1990, 33, 2590 CrossRef CAS.
- B. F. Cain, J. Org. Chem., 1976, 41, 2029 CrossRef CAS.
- S. Milstien and L. A. Cohen, J. Am. Chem. Soc., 1972, 94, 9158 CrossRef.
- L. A. Carpino, S. A. Triolo and R. A. Berglund, J. Org. Chem., 1989, 54, 3303 CrossRef CAS.
- R. Erez and D. Shabat, Org. Biomol. Chem., 2008, 6, 2669 RSC.
- D. Shabat, R. Perry and R. J. Amir, New J. Chem., 2007, 31, 1307 RSC.
- C. Alexander and W. Wang, Angew. Chem., Int. Ed., 2008, 47, 7804 CrossRef.
- D. Shabat and E. Sella, J. Am. Chem. Soc., 2009, 131, 9934 CrossRef CAS.
- D. Shabat and E. Sella, Chem. Commun., 2008, 5701 RSC.
- A. Warnecke and F. Kratz, J. Org. Chem., 2008, 73, 1546 CrossRef CAS.
- D. Shabat, R. Weinstein, A. Sagi and N. Karton, Chem.–Eur. J., 2008, 14, 6857 CrossRef CAS.
- R. Spindler and J. M. J. Fréchet, Macromolecules, 1993, 26, 4809 CrossRef CAS.
- D. Shabat, A. Sagi, R. Weinstein and N. Karton, J. Am. Chem. Soc., 2008, 130, 5434 CrossRef CAS.
- R. Weinstein, P. S. Baran and D. Shabat, Bioconjugate Chem., 2009, 20, 1783 CrossRef.
- A. P. Esser-Kahn, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2010, 132, 10266 CrossRef CAS.
- M. A. Dewit and E. R. Gillies, J. Am. Chem. Soc., 2009, 131, 18327 CrossRef CAS.
- M. A. Dewit, A. Beaton and E. R. Gillies, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3977 CrossRef CAS.
- D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719 CrossRef CAS.
- E. Buhleier, W. Wehner and F. Vögtle, Synthesis, 1978, 78, 155 CrossRef.
- M. A. Naylor, W. A. Goddard III, G. E. Kiefer and D. A. Tomalia, J. Am. Chem. Soc., 1989, 111, 2339 CrossRef.
- C. Hawker and J. M. J. Fréchet, J. Chem. Soc., Chem. Commun., 1990, 1010 RSC.
- M. Gingras, J.-M. Raimundo and Y. M. Chabre, Angew. Chem., Int. Ed., 2007, 46, 1010 CrossRef CAS.
- R. L. McCarley and W. Ong, Macromolecules, 2006, 39, 7295 CrossRef.
- W. Ong and R. L. McCarley, Chem. Commun., 2005, 4699 RSC.
- W. Ong, Y. Yang, A. C. Cruciano and R. L. McCarley, J. Am. Chem. Soc., 2008, 130, 14739 CrossRef CAS.
- D. Shabat, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1569 CrossRef CAS.
- D. Shabat, A. Sagi, E. Segal and R. Satchi-Fainaro, Bioorg. Med. Chem., 2007, 15, 3720 CrossRef CAS.
- L.-C. Lo and C.-Y. Chu, Chem. Commun., 2003, 2728 RSC.
- M. L. Szalai and D. V. McGrath, Tetrahedron, 2004, 60, 7261 CrossRef CAS.
- P. D. Senter, B. E. Toki, C. G. Cerveny and A. F. Wahl, J. Org. Chem., 2002, 67, 1866 CrossRef CAS.
- P. J. Burke, B. E. Toki, D. W. Meyer, J. B. Miyamoto, K. M. Kissler, M. Anderson, P. D. Senter and S. C. Jeffrey, Bioorg. Med. Chem. Lett., 2009, 19, 2650 CrossRef CAS.
- N. Fomina, C. McFearin, M. Sermsakdi, O. Edigin and A. Almutairi, J. Am. Chem. Soc., 2010, 132, 9540 CrossRef CAS.
- K. Haba, M. Popkov, M. Shamis, R. A. Lerner, C. F. Barbas III and D. Shabat, Angew. Chem., Int. Ed., 2005, 44, 716 CrossRef CAS.
- D. Shabat, M. Shamis and H. N. Lode, J. Am. Chem. Soc., 2004, 126, 1726 CrossRef CAS.
- D. Shabat and M. Avital-Shmilovici, Bioorg. Med. Chem. Lett., 2009, 19, 3959 CrossRef CAS.
- D. Shabat and M. Shamis, Chem.–Eur. J., 2007, 13, 4523 CrossRef CAS.
- D. Shabat, R. J. Amir, M. Popkov, R. A. Lerner and C. F. Barbas III, Angew. Chem., Int. Ed., 2005, 44, 4378 CrossRef CAS.
- M. Avital-Shmilovici and D. Shabat, Bioorg. Med. Chem., 2010, 18, 3643 CrossRef CAS.
- E. Sella, A. Lubelski, J. Klafter and D. Shabat, J. Am. Chem. Soc., 2010, 132, 3945 CrossRef CAS.
- E. Sella, R. Weinstain, R. Erez, N. Z. Burns, P. S. Baran and D. Shabat, Chem. Commun., 2010, 46, 6575 RSC.
- M. L. Szalai, S. Li, R. M. Kevwitch and D. V. McGrath, J. Am. Chem. Soc., 2003, 125, 10516 CrossRef CAS.
- D. Shabat, R. J. Amir and E. Danieli, Chem.–Eur. J., 2007, 13, 812 CrossRef CAS.
- D. Shabat, R. Erez, E. Segal, K. Miller and R. Satchi-Fainaro, Bioorg. Med. Chem., 2009, 17, 4327 CrossRef CAS.
- D. Shabat, A. Gopin, S. Ebner and B. Attali, Bioconjugate Chem., 2006, 17, 1432 CrossRef CAS.
- D. Shabat and E. Danieli, Bioorg. Med. Chem., 2007, 15, 7318 CrossRef CAS.
- R. Satchi, T. A. Connors and R. Duncan, Br. J. Cancer, 2001, 85, 1070 CrossRef CAS.
- K. Ulbrich, J. Strohalm, J. Kopecek, B. Rihova, M. Bilej, V. Vetvicka and R. Duncan, Biomaterials, 1989, 10, 335 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688 CrossRef CAS.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387 CAS.
- S.-Q. Gao, Z.-R. Lu, B. Petri, P. Kopeckova and J. Kopecek, J. Controlled Release, 2006, 110, 323 CrossRef CAS.
- S. Sakuma, Z.-R. Lu, P. Kopeckova and J. Kopecek, J. Controlled Release, 2001, 75, 365 CrossRef CAS.
- B.-Y. Ryu, J.-S. Sohn, M. Hess, S.-K. Choi, J.-K. Choi and B.-W. Jo, J. Biomater. Sci., Polym. Ed., 2008, 19, 311 CrossRef CAS.
- R. B. Greenwald, A. Pendri, C. D. Conover, H. Zhao, Y. H. Choe, A. Martinez, K. Shum and S. Guan, J. Med. Chem., 1999, 42, 3657 CrossRef.
- R. B. Greenwald, K. Yang, H. Zhao, C. D. Conover, S. Lee and D. Filpula, Bioconjugate Chem., 2003, 14, 395 CrossRef CAS.
- R. B. Greenwald, Y. H. Choe, C. D. Conover, K. Shum, W. Dechun and M. Royzen, J. Med. Chem., 2000, 43, 475 CrossRef CAS.
- S. Lee, R. B. Greenwald, J. McGuire, K. Yang and C. Shi, Bioconjugate Chem., 2001, 12, 163 CrossRef CAS.
- R. B. Greenwald, H. Zhao, K. Yang, P. Reddy and A. Martinez, J. Med. Chem., 2004, 47, 726 CrossRef CAS.
- H. Zhao, K. Yang, A. Martinez, A. Basu, R. Chintala, H.-C. Liu, A. Janjua, M. Wang and D. Filpula, Bioconjugate Chem., 2006, 17, 341 CrossRef CAS.
- J. W. Suggs and R. M. Pires, Tetrahedron Lett., 1997, 38, 2227 CrossRef CAS.
- R. Satchi-Fainaro, H. Hailu, J. W. Davies, C. Summerford and R. Duncan, Bioconjugate Chem., 2003, 14, 797 CrossRef CAS.
- P. D. Senter, H. P. Svensson, G. J. Schreiber, J. L. Rodriguez and V. M. Vrudhula, Bioconjugate Chem., 1995, 6, 389 CrossRef CAS.
- D. Shabat, R. J. Amir, A. Gopin, N. Pessah and M. Shamis, Chem.–Eur. J., 2004, 10, 2626 CrossRef CAS.
- D. Shabat, A. Gopin, N. Pessah, M. Shamis and C. Rader, Angew. Chem., Int. Ed., 2003, 42, 327 CrossRef CAS.
- D. Shabat, A. Gopin and C. Rader, Bioorg. Med. Chem., 2004, 12, 1853 CrossRef CAS.
- A. Zheng, D. Shan and B. Wang, J. Org. Chem., 1999, 64, 156 CrossRef CAS.
- A. Zheng, D. Shan, X. Shi and B. Wang, J. Org. Chem., 1999, 64, 7459 CrossRef CAS.
- R. Reents, D. A. Jeyaraj and H. Waldmann, DDT, 2002, 7, 71 Search PubMed.
- J.-A. Richard, Y. Meyer, V. Jolivel, M. Massonneau, R. Dumeunier, D. Vaudry, H. Vaudry, P. Y. Renard and A. Romieu, Bioconjugate Chem., 2008, 19, 1707 CrossRef CAS.
- U. Grether and H. Waldmann, Angew. Chem., Int. Ed., 2000, 39, 1629 CrossRef CAS.
- U. Grether and H. Waldmann, Chem.–Eur. J., 2001, 7, 959 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |