Multifunctional and biodegradable polyphosphazenes for use as macromolecular anti-cancer drug carriers†
Ian
Teasdale
*,
Sandra
Wilfert
,
Ivo
Nischang
and
Oliver
Brüggemann
Institute of Polymer Chemistry (ICP), Johannes Kepler University Linz, Welser Str 42, A-4060, Leonding, Austria. E-mail: ian.teasdale@jku.at; Fax: +43 67154762; Tel: +43 67154765
First published on 23rd December 2010
Abstract
Using a living cationic polymerisation procedure we synthesised a series of multi-armed poly(organo)phosphazenes with controlled molecular weights and excellent aqueous solubility. The synthetic flexibility of polyphosphazenes was exploited in order to incorporate an acid-sensitive hydrazide linker to the polymer backbone, as well as tumour-targeting folic acid groups. We were then able to attach hydrophobic anti-cancer drug molecules via the pH labile linker and studied its pH-triggered release kinetics from the polymeric carrier. Although stable for short periods (several days) in an aqueous environment, the polymers were shown to degrade over longer periods (weeks) under simulated physiological conditions. Furthermore, the rate of degradation could be tailored through careful selection of substituents. These biodegradable, multi-functional polyphosphazenes represent promising candidates for use as macromolecular carriers for the tumour-targeted delivery of anti-cancer drugs.
Introduction
Polyphosphazenes have tremendous potential as materials for biomedical applications, offering synthetic flexibility, hydrolytic degradability and non-toxic degradation products.1 The room temperature living cationic polymerisation of chlorophosphoranimine, pioneered by Allcock and Manners,2,3 enables the synthesis of polyphosphazenes with controlled molecular weights and narrow molecular weight distributions, previously unavailable via the classical ring opening polymerisation of hexachlorophosphazene. The development of a living polymerisation route to polyphosphazenes was a key development allowing access to block copolymers,4,5 star-branched and dendritic polymers based on polyphosphazenes.6,7A further property of polyphosphazenes, desirable for many biomedical applications, is the inherent hydrolytic instability of the polyphosphazene backbone. The hydrolytic stability can vary greatly, depending on the properties of the side-substituents and hydrophilicity of the polymer.8,9 This can be readily utilised to give a broad spectrum of polymers with very different rates of degradation. In addition to the corresponding side groups, polyphosphazenes have been shown to degrade to low toxicity compounds including ammonia and phosphates.10 In particular, hydrophilic amino substituted polyphosphazenes are known to be hydrolytically unstable and the stability can be tailored by careful choice of substituents such as depsipeptides, carboxylic acids or amino acid esters.10–18
The biocompatibility of poly(organo)phosphazenes has led to their development in numerous biomedical applications, including hydrogel scaffolds for tissue engineering,19–21polyelectrolytes for use as vaccine immunoadjuvants22,23 as polyplexes for gene delivery24 and as imaging agents.25 A number of authors have also reported their use in drug delivery applications, in particular as degradable matrices,17,26 and including in vivo studies on the local delivery of anti-cancer drugs from polyphosphazene hydrogels27–29 and micelles.30,31 Polyphosphazenes have also been investigated as carriers of the anti-cancer drug cisplatin32,33 and some other platinum-based anti-cancer agents.34–36
In this work we demonstrate how the adjustable biodegradability and synthetic flexibility of polyphosphazenes can be exploited, in combination with the recently developed controlled living polymerisation, in order to design complete macromolecular drug carriers, which, as well as being capable of loading anti-cancer drugs and targeting ligands, would have good aqueous solubility, controlled molecular weights, narrow polydispersities, rapid pH-activated drug-release and biodegradability.
Macromolecular drug carriers have been shown to improve the therapeutic index of anti-cancer drug molecules and to improve their blood solubility and circulation time.37–40 Control of molecular weight and dispersity is an important factor as molecular architecture and hydrodynamic volume of the polymer play a crucial role in the pharmacokinetics and in vivo distribution of polymeric drug carriers.41Macromolecules with hydrodynamic volumes above a certain threshold lead to reduced renal clearance, thus increasing blood circulation time. Meanwhile, the enhanced permeation and retention effect (EPR) can cause accumulation of larger macromolecules in tumour tissue and thus lead to passive tumour targeting been shown to increase with increasing polymer size.42 However, polymers with hydrodynamic volumes above the renal filtration limit cannot subsequently be removed from the body and therefore biodegradable polymers are desirable in order to avoid undesired longer term accumulation of the polymer in the body.39,42 Alongside the EPR effect, tumour targeting can also be achieved through attachment targeting moiety onto the macromolecular carriers.38,43
Targeted release of drug payloads from macromolecular carriers can be achieved through attachment of the load via a cleavable linker.38,44,45 The pH of tumour tissue is reported to be lower than that of healthy tissue, thus lending itself to the use of acid-degradable linkages.45–48 Using an acid-cleavable hydrazide linker, we investigate the loading and the subsequent pH-triggered release of the anthracycline antibiotics, epirubicin and doxorubicin, which have well-reported anti-tumour activity.49,50
Experimental
Materials
All solvents were dried using standard laboratory procedures. All synthetic procedures were carried out either in a glove box (MBRAUN) under argon or under nitrogen using standard Schlenk line techniques. Epirubicin hydrochloride was purchased from Molekula Deutschland Ltd. (Taufkirchen, Germany). Amine capped polyetheramine copolymers (PEO–PPO–NH2), sold under the trade name Jeffamines, were donated by Huntsman Performance Products and used as received. Unless otherwise stated, the PEO–PPO–NH2 had an Mn of 1000 and an ethylene oxide/propylene oxide ratio of 19/3. The polyetheramine with an Mn of 2070 had an ethylene oxide/propylene oxide ratio of 31/10. PCl5 was purified by sublimation and stored under argon. Triethylamine was dried over molecular sieves and distilled prior to use. All other chemicals were purchased from Sigma Aldrich and used without prior purification. All glassware was dried in an oven overnight prior to use.Characterisation
Characterisation by NMR spectroscopy was conducted on Bruker 200 MHz spectrometer using CDCl3, DMSO-d6 or D2O, as reported. 31P NMR (81 MHz) was conducted using 85% phosphoric acid as an external standard. UV-Vis spectra were carried out on a Perkin Elmer Lambda 25 UV/VIS spectrophotometer. Gel permeation chromatography was carried out on a Viscotek HT-GPC instrument using two PLgel mixed bead columns assembled in series and a refractive index detector. Molecular weights were estimated from Viscotek Polycal polystyrene standards. Samples were eluted at 35 °C with THF containing 0.1% (w/w) tetra-n-butyl ammonium nitrate. FTIR spectra were measured with a Perkin Elmer Spectrum 100 FTIR spectrometer. A 1290 Infinity UPLC system (Agilent Technologies, Vienna, Austria) equipped with a diode array detector and a Zorbax Eclipse Plus C18 column (2.1 mm × 50 mm, 1.8 µm particle size) was used for kinetic studies of the drug release. The samples were eluted at a flow rate of 0.5 mL min−1 at room temperature with a mobile phase composition of 25% acetonitrile in water (v/v) containing 0.1% formic acid (v/v) in isocratic mode. UV detection was carried out at 254 nm in the linear range of the detector.Synthesis of Cl3PNSiMe351
40 g LiN(SiMe3)2 (239 mmol) were dissolved in 800 mL diethylether. The reaction was then cooled to 0 °C and stirred for 30 min. 20.91 mL PCl3 (239 mmol) were then added dropwise at 0 °C. The solution was allowed to warm to room temperature and stirred for 2.5 hours. After cooling to 0 °C again, 19.35 mL SO2Cl2 (239 mmol) were added and the mixture was stirred for another 3 hours at 0 °C. The reaction was filtered through Celite and the volatiles removed under vacuum. The product was purified by vacuum distillation (50 °C, 4 mbar) to yield chlorophosphoranimine as a colourless, viscous oil. The product was stored under inert argon atmosphere at −40 °C. Yield 35%; 1H NMR (CDCl3): δ = 0.15 (s, 9H) ppm, 31P NMR (CDCl3); −54.1 ppm.Synthesis of β-alanyl-boc-hydrazide
The boc-protected linker, β-alanyl-boc-hydrazide was synthesised similar to literature procedures.52Z-β-Ala-OH (5.00 g, 22.4 mmol), boc-NH-NH2 (2.96 g, 22.4 mmol) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI) (4.51 g, 23.51 mmol) were dissolved in 200 mL CH2Cl2 and stirred for 2 hours at room temperature. The reaction mixture was extracted with 200 mL of 0.1 M acetic acid. The aqueous layer was extracted three times with 50 mL CH2Cl2. The organic layers were then combined and extracted twice with 200 mL 0.1 M acetic acid, twice with 200 mL of saturated aqueous sodium hydrogen carbonate and once with 200 mL H2O. The organic layer was dried over MgSO4, solvents removed under vacuum and the product then further dried under high vacuum to yield Z-β-alanyl-boc-hydrazide as a white powder. The product (5.01 g, 14.86 mmol) was hydrogenated at 3 bar in 150 mL methanol with 10% Pd–C (0.3 g) for 24 hours. The reaction was filtered through Celite and rotary evaporated. The product was dried under high vacuum to yield β-alanyl-boc-hydrazide as a white foam. Yield 65%, FTIR (solid) ν max/cm−1 = 3259br (N–H), 2977w (C–H), 1671s (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (CDCl3): δ = 1.46 (s, 9H), 2.50 (m, 2H), 3.13 (m, 2H), 5.30 (br, 4H) ppm.
O). 1H NMR (CDCl3): δ = 1.46 (s, 9H), 2.50 (m, 2H), 3.13 (m, 2H), 5.30 (br, 4H) ppm.
Synthesis of pegylated folic acid
Di-tert-butyl carbonate (0.9 g, 0.66 mmol) was added dropwise to a solution of o,o′-bis(3-aminopropyl)polyethylene glycol (144 mg, 0.60 mmol) in dioxane (20 mL) and triethylamine (73 mg, 0.70 mmol). The mixture was then stirred at room temperature for 16 hours. The solvent was removed under vacuum, the product dissolved in CH2Cl2 and reprecipitated into diethylether/hexane at −15 °C. The white solid was filtered and used for the next step. A portion of the product (320 mg, 0.21 mmol) and EDCI (45 mg, 0.23 mmol) were added to a flask and placed under nitrogen. In a separate vessel, folic acid (103 mg, 0.23 mmol) was dissolved with heating in DMF (25 mL). The solution was returned to room temperature and added to the reaction mixture. The mixture was stirred for 12 hours at room temperature. The DMF was removed under high vacuum and the product purified in a PD-10 Sephadex column, eluted with 0.1 M NaHCO3. The product was then deprotected in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2:CF3COOH solution and stirred for 3 hours at room temperature after which the solvent was removed under high vacuum. Overall yield 35%. 1H NMR (DMSO-d6): δ = 1.69 (m, 4H), 1.92 (br, 2H), 2.12 (br, 2H), 2.74 (t, 4H), 3.14 (m, 2H), 3.50 (br, 136H), 3.85 (m, 2H), 4.08 (m, 1H), 4.43 (br, 2H), 6.63 (d, 2H), 6.83 (s, 2H), 6.59 (d, 2H), 7.93 (s, 1H), 8.57 (s, 1H) ppm. UV-Visλ max (0.1 M NaOH)/nm 256, 283 and 368 (ε/dm3 mol−1 cm−1 26900, 21500 and 9120).
:1 CH2Cl2:CF3COOH solution and stirred for 3 hours at room temperature after which the solvent was removed under high vacuum. Overall yield 35%. 1H NMR (DMSO-d6): δ = 1.69 (m, 4H), 1.92 (br, 2H), 2.12 (br, 2H), 2.74 (t, 4H), 3.14 (m, 2H), 3.50 (br, 136H), 3.85 (m, 2H), 4.08 (m, 1H), 4.43 (br, 2H), 6.63 (d, 2H), 6.83 (s, 2H), 6.59 (d, 2H), 7.93 (s, 1H), 8.57 (s, 1H) ppm. UV-Visλ max (0.1 M NaOH)/nm 256, 283 and 368 (ε/dm3 mol−1 cm−1 26900, 21500 and 9120).
Polymerisation procedure
Polymers were synthesised according to Allcocks' procedure for the living cationic polymerisation of chlorophosphoranimine.3 The following example procedure describes the procedure used for the synthesis of polymer 1. Other polymers were synthesised accordingly, with the ratio of monomer to initiator varied and the relative amounts of substituents adjusted in order to obtain the desired polymers.In the glove box, initiator PCl5 (18.55 mg, 0.09 mmol) and monomer Cl3PNSiMe3 (0.51 g, 2.26 mmol) were dissolved in CH2Cl2 (5 mL) at room temperature. The solution was stirred for 12 h and the solvent removed under vacuum. The resulting polydichlorophosphazene was then dissolved in anhydrous THF in an inert atmosphere. 0.4 equivalent of the hydrazide linker (0.18 g, 0.91 mmol) and NEt3 (0.09 g, 0.91 mmol) were then added to the polymer solution and allowed to react for 24 hours. An excess of PEO–PPO–NH2 (2.6 eq, 5.89 g, 5.89 mmol) was then added to the reaction mixture and allowed to react for a further 24 hours. The solvent was then removed under vacuum and resulting polymers were purified by dialysis (12 kDa cut-off) for 48 hours against deionized H2O followed by 24 hours against MeOH. The solvent was removed under a stream of nitrogen and the polymers were dried under vacuum to give waxy solids or highly viscous liquids in yields of 50–60%. All polymers were analysed by GPC analysis, 31P NMR, 1H NMR and FTIR spectroscopy (ESI†). All other polymers were synthesized using this procedure with the exception that polymer 5 was synthesised via the sequential addition of 0.25 eq linker, 0.01 eq FA–PEO–NH2 and then an excess (2.74 eq) PEO–PPO–NH2 and that polymers 6–8 were synthesised with 0.2 eq linker, followed by the desired amount of PEO–PPO–NH2 and then an excess of ethyl glycinate ester, with 24 hours reaction time allowed between each addition.
Loading of anti-cancer drugs
A sample of the protected polymer (150 mg) was dissolved in a 2:1 CH2Cl2:CF3COOH solution and stirred for 3 hours. The solvent was then removed under high vacuum. The deprotected polymer was then added to anhydrous methanol (10 mL) and 1 equivalent per hydrazide group of epirubicin hydrochloride. The mixture was stirred under reflux for 24 hours. The product was then purified by dialysis against methanol for 5 days. The amount of epirubicin hydrochloride covalently bound to the polymers was measured in H2O by UV-Vis spectroscopy from the absorbance at 481 nm (ε = 11200).53 The identical procedure was also used to load doxorubicin hydrochloride onto the polymers, with the amount bound to the polymers being measured from the absorbance at 488 nm (ε = 11500).47
Polymer degradation studies
Polymer samples (0.20 g) were dissolved in a pH 7.4 phosphate buffer solution and incubated at 37 °C. An aliquot (0.25 mL) was then removed at regular intervals and the solvent was evaporated. The polymer was then dissolved in THF, filtered through a 45 µm PTFE filter and analysed by GPC with an RI detector.Results and discussion
The room temperature, cationic living polymerisation of chlorophosphoranimine provided a route to a series of dichloropolyphosphazenes with narrow polydispersities (Scheme 1). A boc-protected hydrazide linker, for subsequent drug attachment, and an amine-capped polyalkylene oxide copolymer (PEO–PPO–NH2) were chosen as side-group substituents to replace the chlorine atoms (Scheme 2). Due to the hydrophilicity provided by the PEO–PPO–NH2 side chains, the series of polymers showed excellent aqueous solubility (with the exception of polymer 3, with only 5% PEO–PPO side chains). 31P NMR analysis confirmed that all chlorine atoms have been substituted, but only one broad peak was observed due to the random, mixed geminal substitution pattern.54 The relative ratios of the two substituents could, however, be calculated by 1H NMR spectroscopy, through integration of the PEO protons at 3.6 ppm versus those from the boc groups of the hydrazide linker at 1.4 ppm (Table 1). Although the molecular weights of the polymers could be carefully controlled by varying the initial ratio of initiator to monomer, as well as the subsequent substituents, the molecular weights measured by GPC and calibrated against linear polystyrene standards were a factor of 2–3 lower than that estimated by the initiator:monomer ratio, with the factor greater for polymers with a higher molecular weight (Table 1). This deferred elution time is attributed to the branched, closely packed nature of the polymers, leading to a lower hydrodynamic volume in comparison to the linear standards.55 It should also be noted that the relative molecular weights of linear polyphosphazenes, as determined by polystyrene standards, are also often observed to differ from their absolute weights.3,56 This, along with the branched nature of the polymers, means that the molecular weights as measured by GPC can only be considered as a guide to the absolute molecular weights. The polydispersities Mw/Mn were measured to be 1.2–1.5. This is slightly higher than the usual range for these polymers (1–1.3)57 and is thought to be a consequence of the mixed substitution of the side chains, which would be expected to produce a statistical distribution of substituents.
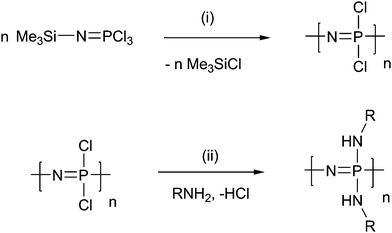 | ||
| Scheme 1 Living polymerisation of poly(organo)phosphazenes. Reagents and conditions: (i) PCl5, CH2Cl2, RT; (ii) NEt3, THF, RT. | ||
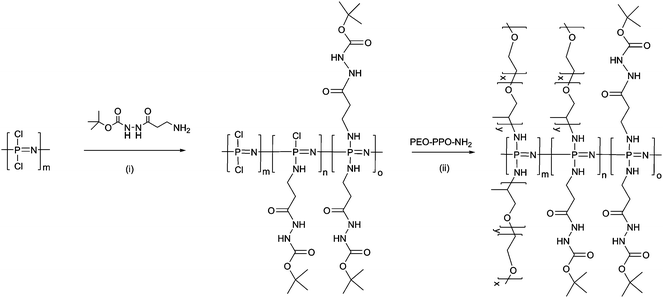 | ||
| Scheme 2 Synthesis of polymers 1–4, hydrophilic polyphosphazene copolymers with a boc-protected hydrazide linker and hydrophilic polyalkylene oxide side chains. Reagents and conditions: (i) and (ii) THF, NEt3 RT, 24 h. | ||
| Polymer | M:I |
Linker/PEO–PPO ratioa | M n(calc) b/kg mol−1 | M n c/kg mol−1 | M w/ M n |
|---|---|---|---|---|---|
|
a Measured by 1H NMR.
b Calculated from the initial monomer:initiator and side group ratios.
c Measured by GPC analysis and calibrated against linear polystyrene standards.
d Synthesised with 2070 Mn PEO–PPO–NH2 side chains.
|
|||||
| 1 | 25:1 |
1:2 |
80 | 32 | 1.27 |
| 2 | 25:1 |
1:1.7 |
83 | 31 | 1.47 |
| 3 | 25:1 |
1:0.1 |
27 | 14 | 1.30 |
| 4 d | 25:1 |
1:1 |
159 | 49 | 1.30 |
| 5 | 50:1 |
1:2 |
160 | 51 | 1.44 |
The folate receptor has been shown to be over-expressed in many human cancers43 and its conjugation to macromolecular carriers has been successfully implemented by a number of authors.43,58–60 To this end, a small amount of a folic acid-capped amino polyalkylene oxide was added to polymer 5 and UV-Vis spectroscopy was used to confirm the incorporation of folic acid moieties in the polymers (Fig. 1), which showed a loading of 0.5 wt% of the pegylated folic acid, an average of 1–2 folic acid moieties per macromolecule. The coupling of folic acid to the macromolecules demonstrates the synthetic flexibility of these macromolecular drug carriers and it is anticipated that alternative tumour-targeting ligands61 (e.g.biotin) could be simply attached to the polymeric carriers in a similar manner.
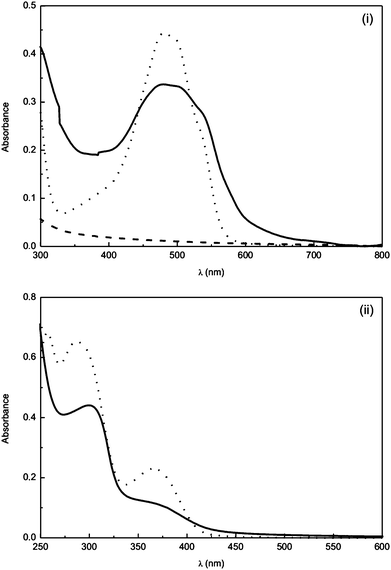 | ||
| Fig. 1 UV-Vis spectra in H2O of (i) epirubicin (dotted line) and polyphosphazene 1 (dashed line) and polyphosphazene 1 loaded with 2 wt% epirubicin hydrochloride (solid line); (ii) folic acid (dotted line) and polyphosphazene 5, loaded with 0.5 wt% folic acid. | ||
Following deprotection of the hydrazide groups, anti-cancer drugs could be successfully loaded onto the polymersvia reaction of the resulting amine groups with the carbonyl group in the side chain of epirubicin (or its stereoisomer doxorubicin) (Scheme 3). Successful loading of the drug was confirmed and the percentage loading calculated by UV-Vis analysis (Fig. 1) from the absorbance at 481 nm. Loading was, however, lower than expected (∼2 to 3%), with only approximately 10% of the total available hydrazide groups bearing drug moieties. Further reactions, in which the polymer–drug solution was heated to reflux did improve the loading and enabled the preparation of polymers with a loading of up to 7%, which corresponds to approximately 40% of the total hydrazide moieties.
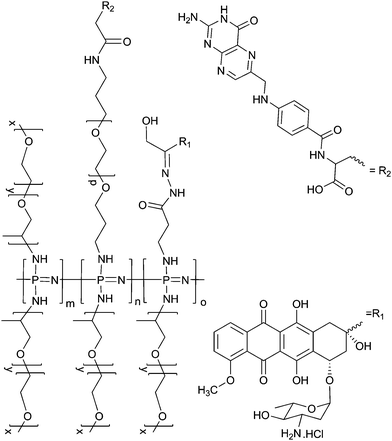 | ||
| Scheme 3 Simplified structure for hydrophilic poly (organo)phosphazenes loaded with epirubicin and folic acid moieties. Combinations of the three different side chains are expected to be statistically distributed. | ||
The release of epirubicin from the polymer–drug conjugates was then analysed by HPLC under simulated physiological conditions at 37 °C at pH 7.4 and at pH 5 (Fig. 2). At pH 5 a steady release of the drug molecule from the polymer was observed, with 100% release from the polymer–drug conjugate being observed within 15 hours. Meanwhile, only minimal release was observed within a period of 24 h from the polymers at pH 7.4. The rate of release is comparable to reports from authors using similar hydrazide based polymer systems.39,62
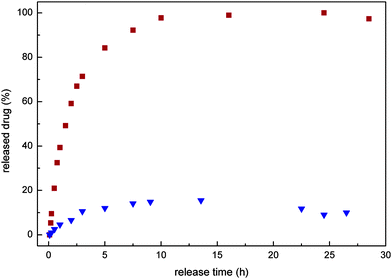 | ||
| Fig. 2 Release of epirubicin from the hydrazone-linked polyphosphazene 1 at 37 °C in acidic environment ■ (acetate buffer, pH 5), and a neutral solution▼ (pH 7.4, phosphate buffer). The amount of the released epirubicin was estimated using a calibration curve for the free drug. | ||
The biodegradability of a selection of these polymers (tested at 37 °C and pH 7.4), monitored by size exclusion chromatography, showed that the polymers are stable over a short period of time in an aqueous environment, making these viable candidates for the intended drug-delivery application. The polymers did, however, undergo degradation over longer periods under these simulated physiological conditions (Fig. 3). A clear broadening and a shift to longer retention time of the polymer peak are observed. Furthermore, a peak in the GPC chromatographs was observed to appear at a later retention time. This peak, which increases in relative intensity over time, corresponds to an Mn ≈ 1000, strongly suggesting that the polyalkyleneoxide side chains are first ejected from the polymers. This supports previous studies which suggest that the mechanism for degradation of amino substituted polyphosphazenes involves removal of the side groups to form hydroxyphosphazenes and phosphazanes, which then undergo rapid hydrolytic chain cleavage.8
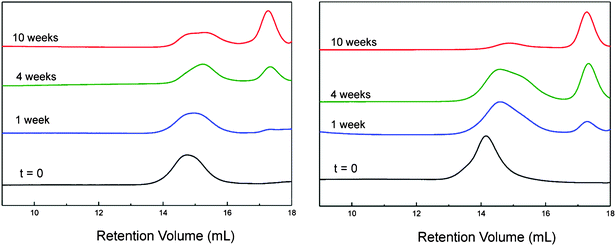 | ||
| Fig. 3 GPC chromatographs showing the degradation of polymer 2 (left) and polymer 6 (right) at 37 °C in an aqueous buffer solution (pH 7.4). Broadening and decrease in intensity and a shift to longer retention time of the polymer peak are observed alongside an increase in the peak associated with the polyalkylene oxide side chains as they are eliminated from the polymer. Polymer 6, incorporating 25% ethyl glycinate side groups, degrades considerably faster than for polymer 2, with no amino acid ester side groups. | ||
Although the polymers need to be robust to act as drug carriers (for the loading and delivery processes), polymers which degrade too slowly will be expected to accumulate in the body. The ability to delay or accelerate the degradation is limited with other polymeric systems63 and the facile tailoring is potentially a significant advantage for polyphosphazene based systems, the rate of degradation of which can be altered significantly by careful choice of substituents. In particular, the incorporation of amino acid side chains has been shown to considerably decrease the hydrolytic stability of hydrophilic poly(organo)phosphazenes.11,64 To this end, we synthesised a series of polymersvia sequential addition of linker, PEO–PPO–NH2 and then ethyl glycinate ester side chains in varying ratios (Table 2). The polymers all showed good water solubility but the incorporation of a third different side chain led to a small increase in the polydispersity. As shown in Fig. 3, the degradation is considerably accelerated upon incorporation of ethyl glycinate side groups. After 2 weeks, the Mn of polymer 6, in which around 47% of the chlorine atoms were substituted with ethyl glycinate groups, was reduced to 66% of its original value, whereby polymer 2 had an Mn value 80% of its original. The effect of polymer molecular weight, hydrophilicity, of steric crowding, as well as the different ratios of polyalkylene oxide and amino acid ester groups render a direct correlation for the rate of degradation for all polymers difficult and the precise impact of these relationships requires further investigation8,11 in order to attain a better understanding and hence ability to truly tailor the precise required rate of degradation.
Conclusions
These results demonstrate the potential of polyphosphazenes to create defined and tunable macromolecules for the targeted delivery of anti-cancer drugs. We have described the synthesis of hydrophilic, biodegradable polyphosphazenes via the living polymerisation procedure. The polymers can be readily decorated with a combination of targeting ligands and drug molecules. We have also successfully demonstrated the pH-triggered release of an anti-cancer drug from the polymer–drug conjugates, as well as the tailoring of the biodegradability by incorporation of amino acid ester side groups. These polymers represent extremely promising candidates for the targeted delivery of anti-cancer drugs.Acknowledgements
The authors would like to thank Mrs Ramona Kiss and Dr Mario Waser for their assistance and Huntsman Performance Products for their donation of Jeffamines used in this study.References
- Polyphosphazenes for Biomedical Applications, ed. A. K. Andrianov, Wiley, Hoboken, 2009 Search PubMed.
- V. Blackstone, A. J. Lough, M. Murray and I. Manners, J. Am. Chem. Soc., 2009, 131, 3658–3667 CrossRef CAS.
- H. R. Allcock, C. A. Crane, C. T. Morrissey, J. M. Nelson, S. D. Reeves, C. H. Honeyman and I. Manners, Macromolecules, 1996, 29, 7740–7747 CrossRef CAS.
- J. M. Nelson, A. P. Primrose, T. J. Hartle, H. R. Allcock and I. Manners, Macromolecules, 1998, 31, 947–949 CrossRef CAS.
- K. Matyjaszewski, M. K. Moore and M. L. White, Macromolecules, 1993, 26, 6741–6748 CrossRef CAS.
- J. M. Nelson and H. R. Allcock, Macromolecules, 1997, 30, 1854–1856 CrossRef CAS.
- S. Y. Cho and H. R. Allcock, Macromolecules, 2007, 40, 3115–3121 CrossRef CAS.
- H. R. Allcock, T. J. Fuller, D. P. Mack, K. Matsumura and K. M. Smeltz, Macromolecules, 1977, 10, 824–830 CrossRef CAS.
- S. E. M. Ibim, A. M. A. Ambrosio, M. S. Kwon, S. F. El-Amin, H. R. Allcock and C. T. Laurencin, Biomaterials, 1997, 18, 1565–1569 CAS.
- H. R. Allcock, S. R. Pucher and A. G. Scopelianos, Biomaterials, 1994, 15, 563–569 CrossRef CAS.
- J. Vandorpe and E. Schacht, Polymer, 1996, 37, 3141–3145 CrossRef CAS.
- H. R. Allcock, S. R. Pucher and A. G. Scopelianos, Macromolecules, 1994, 27, 1071–1075 CrossRef CAS.
- A. Singh, N. R. Krogman, S. Sethuraman, L. S. Nair, J. L. Sturgeon, P. W. Brown, C. T. Laurencin and H. R. Allcock, Biomacromolecules, 2006, 7, 914–918 CrossRef CAS.
- S. Sethuraman, L. S. Nair, S. El-Amin, R. Farrar, M. T. N. Nguyen, A. Singh, H. R. Allcock, Y. E. Greish, P. W. Brown and C. T. Laurencin, J. Biomed. Mater. Res., Part A, 2006, 77, 679–687 CrossRef.
- J. H. L. Crommen, E. H. Schacht and E. H. G. Mense, Biomaterials, 1992, 13, 511–520 CrossRef CAS.
- J. H. L. Crommen, E. H. Schacht and E. H. G. Mense, Biomaterials, 1992, 13, 601–611 CrossRef CAS.
- S. M. Ibim, A. A. Ambrosio, D. Larrier, H. R. Allcock and C. T. Laurencin, J. Controlled Release, 1996, 40, 31–39 CrossRef CAS.
- D. P. DeCollibus, A. Marin and A. K. Andrianov, Biomacromolecules, 2010, 11, 2033–2038 CrossRef CAS.
- S. Sethuraman, L. S. Nair, S. El-Amin, M. T. Nguyen, A. Singh, N. Krogman, Y. E. Greish, H. R. Allcock, P. W. Brown and C. T. Laurencin, Acta Biomater., 2010, 6, 1931–1937 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- S. F. El-Amin, M. S. Kwon, T. Starnes, H. R. Allcock and C. T. Laurencin, J. Inorg. Organomet. Polym. Mater., 2006, 16, 387–396 CAS.
- A. K. Andrianov, A. Marin and B. E. Roberts, Biomacromolecules, 2005, 6, 1375–1379 CrossRef CAS.
- A. K. Andrianov, J. Inorg. Organomet. Polym. Mater., 2006, 16, 397–406 CAS.
- J. Luten, M. J. van Steenbergen, M. C. Lok, A. M. de Graaff, C. F. van Nostrum, H. Talsma and W. E. Hennink, Eur. J. Pharm. Sci., 2008, 33, 241–251 CrossRef CAS.
- A. L. Weikel, S. Y. Cho, N. L. Morozowich, L. S. Nair, C. T. Laurencin and H. R. Allcock, Polym. Chem., 2010, 1, 1459–1466 RSC.
- S. Lakshmi, D. S. Katti and C. T. Laurencin, Adv. Drug Delivery Rev., 2003, 55, 467–482 CrossRef CAS.
- C. Chun, S. M. Lee, C. W. Kim, K. Y. Hong, S. Y. Kim, H. K. Yang and S. C. Song, Biomaterials, 2009, 30, 4752–4762 CrossRef CAS.
- G. D. Kang, S. H. Cheon and S. C. Song, Int. J. Pharm., 2006, 319, 29–36 CrossRef CAS.
- J. Y. Seong, Y. J. Jun, B. M. Kim, Y. M. Park and Y. S. Sohn, Int. J. Pharm., 2006, 314, 90–96 CrossRef CAS.
- J. X. Zhang, L. Y. Qiu, Y. Jin and K. J. Zhu, J. Biomed. Mater. Res., Part A, 2006, 76, 773–780 CrossRef.
- L. Y. Qiu and M. Q. Yan, Acta Biomater., 2009, 5, 2132–2141 CrossRef CAS.
- Y. S. Sohn, H. Baek, Y. H. Cho, Y. A. Lee, O. S. Jung, C. O. Lee and Y. S. Kim, Int. J. Pharm., 1997, 153, 79–91 CrossRef CAS.
- R. Song, Y. J. Jun, J. I. Kim, C. Jin and Y. S. Sohn, J. Controlled Release, 2005, 105, 142–150 CrossRef CAS.
- H. R. Allcock, R. W. Allen and J. P. Obrien, J. Chem. Soc., Chem. Commun., 1976, 717–718 RSC.
- J. P. Obrien, R. W. Allen and H. R. Allcock, Inorg. Chem., 1979, 18, 2230–2235 CrossRef CAS.
- H. R. Allcock, R. W. Allen and J. P. Obrien, J. Am. Chem. Soc., 1977, 99, 3984–3987 CrossRef CAS.
- F. Greco and M. J. Vicent, Adv. Drug Delivery Rev., 2009, 61, 1203–1213 CrossRef CAS.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS.
- C. C. Lee, E. R. Gillies, M. E. Fox, S. J. Guillaudeu, J. M. J. Fréchet, E. E. Dy and F. C. Szoka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16649–16654 CrossRef CAS.
- T. Lammers, V. Subr, K. Ulbrich, W. E. Hennink, G. Storm and F. Kiessling, Nano Today, 2010, 5, 197–212 CrossRef CAS.
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- Y. J. Lu and P. S. Low, Adv. Drug Delivery Rev., 2002, 54, 675–693 CrossRef CAS.
- M. Kramer, J. F. Stumbe, H. Turk, S. Krause, A. Komp, L. Delineau, S. Prokhorova, H. Kautz and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 4252–4256 CrossRef CAS.
- E. R. Gillies and J. M. J. Fréchet, Pure Appl. Chem., 2004, 76, 1295–1307 CrossRef CAS.
- R. Tong and J. J. Cheng, Polym. Rev. (Philadelphia, PA, U. S.), 2007, 47, 345–381 Search PubMed.
- T. Etrych, P. Chytil, M. Jelinkova, B. Rihova and K. Ulbrich, Macromol. Biosci., 2002, 2, 43–52 CrossRef CAS.
- O. L. Padilla De Jesus, H. R. Ihre, L. Gagne, J. M. J. Frechet and F. C. Szoka, Jr, Bioconjugate Chem., 2002, 13, 453–461 CrossRef CAS.
- M. Shin, H. Matsunaga and K. Fujiwara, Histochem. Cell Biol., 2010, 133, 677–682 CrossRef CAS.
- J. W. Lown, Pharmacol. Ther., 1993, 60, 185–214 CrossRef CAS.
- C. H. Honeyman, A. J. Lough and I. Manners, Inorg. Chem., 1994, 33, 2988–2993 CrossRef CAS.
- H. D. King, D. Yurgaitis, D. Willner, R. A. Firestone, M. B. Yang, S. J. Lasch, K. E. Hellstrom and P. A. Trail, Bioconjugate Chem., 1999, 10, 279–288 CrossRef CAS.
- N. Erdinc, S. Gokturk and W. Tuncay, J. Pharm. Sci., 2004, 93, 1566–1576 CrossRef CAS.
- M. D. Hindenlang, A. A. Soudakov, G. H. Imler, C. T. Laurencin, L. S. Nair and H. R. Allcock, Polym. Chem., 2010, 1, 1467–1474 RSC.
- N. Kaskhedikar, J. Paulsdorf, M. Burjanadze, Y. Karatas, D. Wilmer, B. Roling and H. D. Wiemhofer, Solid State Ionics, 2006, 177, 703–707 CrossRef CAS.
- A. K. Andrianov and M. P. Le Golvan, J. Appl. Polym. Sci., 1996, 60, 2289–2295 CrossRef CAS.
- H. R. Allcock, S. D. Reeves, J. M. Nelson, C. A. Crane and I. Manners, Macromolecules, 1997, 30, 2213–2215 CrossRef CAS.
- Y. Q. Zhang, Y. H. Sun, X. P. Xu, X. Z. Zhang, H. Zhu, L. L. Huang, Y. J. Qi and Y. M. Shen, J. Med. Chem., 2010, 53, 3262–3272 CrossRef CAS.
- D. Pan, J. L. Turner and K. L. Wooley, Chem. Commun., 2003, 2400–2401 RSC.
- Y. H. Zhang, T. P. Thomas, A. Desai, H. Zong, P. R. Leroueil, I. J. Majoros and J. R. Baker, Bioconjugate Chem., 2010, 21, 489–495 CrossRef CAS.
- I. Ojima, Acc. Chem. Res., 2008, 41, 108–119 CrossRef CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS.
- D. G. van der Poll, H. M. Kieler-Ferguson, W. C. Floyd, S. J. Guillaudeu, K. Jerger, F. C. Szoka and J. M. Frechet, Bioconjugate Chem., 2010, 21, 764–773 CrossRef CAS.
- A. K. Andrianov and A. Marin, Biomacromolecules, 2006, 7, 1581–1586 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional polymer characterisation data and example spectra. See DOI: 10.1039/c0py00321b |
| This journal is © The Royal Society of Chemistry 2011 |