Shedding the hydrophilic mantle of polymersomes†
René P.
Brinkhuis
*,
Taco R.
Visser
,
Floris P. J. T.
Rutjes
and
Jan C. M.
van Hest
Radboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands. E-mail: f.rutjes@science.ru.nl; j.vanhest@science.ru.nl; Fax: +31 243653393; Tel: +31 243653204
First published on 16th November 2010
Abstract
Block copolymers of polybutadiene-b-poly(ethylene glycol) were prepared in which both segments were coupled via an acid sensitive hydrazone moiety. Polymersomes that were subsequently formed showed a strong pH-dependent colloidal stability as a result of the pH sensitive removal of the PEG block. By mixing this stimulus responsive block polymer with an inert analogue it was possible to systematically remove percentages of PEG from the polymersome mantle. The minimum amount of surface PEGylation needed to retain stable polymersomes was found to be as low as five percent.
Polymeric vesicles, or polymersomes, can be regarded as the polymeric analogues of liposomes. They are formed by the self-assembly of amphiphilic block copolymers in aqueous media.1,2 Compared to liposomes, polymersomes are characterized by an increased membrane stability and the ability to enclose larger quantities of hydrophobic compounds.2 This makes them highly interesting for use as nanocarriers in biomedical applications such as drug delivery and in vivo imaging.3 Nowadays researchers have gained a high degree of control over polymersome composition, size and peripheral functionalities which are all important elements in the design of drug delivery vehicles.4 An important challenge, however, remains the triggered release of compounds from the interior of the polymersomes, which requires a controlled destabilization of the membrane.
Destabilization of the particles is often realized by degradation or a triggered change in solubility of one of the blocks,5 but it can in principle also be realized by removal of the stabilizing hydrophilic segment of the amphiphilic block copolymer. This hydrophilic block is in most cases the biocompatible polymer poly(ethylene glycol) (PEG) which introduces stealth-like properties to the nanocarriers when they circulate through the body. Basically it is the shielding of the hydrophobic domain by highly stretched peripheral PEG chains6 which gives rise to the stability of polymersomes in water. Until now it is, however, unclear how much PEG is required to maintain a stable colloidal polymeric capsule. To investigate this, a systematic removal of different percentages of PEG from the polymersome surface should be accomplished. This requires the introduction of a triggerable cleavage site between the hydrophobic and hydrophilic parts of the block copolymer that constitutes the polymersome membrane.
Different block copolymer cleavage methods have already been reported, based on UV light,7,8 reductive9–14 or oxidative and enzymatic pathways.15 Site selective acidic cleavable block copolymers reported today make use of a triphenyl ether linker,16 a cyclic ortho ester17 or cis-aconitic acid.18 All other acid labile systems are based on random hydrolysis.19–22 An interesting pH cleavable moiety for block copolymer cleavage is the hydrazone linkage. This functionality is well known for its strong acid dependent stability in the physiological pH range.23–25Hydrazones have been used in the field of liposomal delivery systems as cleavable linkers between PEG chains and surface peptides in order to hide the peptides up to the moment of cellular uptake.26 In another example Kataoka et al. showed the efficient release of drugs, conjugated viahydrazone bonds, from polymeric micelles upon lowering the pH to endosomal levels.27 Amphiphilic block copolymers of which the hydrophobic and hydrophilic parts are connected by the pH sensitive hydrazone linkage have very recently been reported to form pH responsive aggregates in aqueous solution.28
In this paper the controlled colloidal destabilization of polymersomes formed by the self-assembly of amphiphilic block copolymers of polybutadiene-b-poly(ethylene glycol) (pBd–PEG) is investigated. By using different combinations of an inert amphiphilic block copolymer and pBd–PEG of which both parts are connected by a hydrazone linker, the minimum amount of PEG needed for stabilization is determined. We show that it is possible to take away 95 percent of peripheral PEG chains by lowering the pH, without disrupting the colloidal stability of polymersomes. Furthermore, we can tune the speed of polymersome degradation by adjusting the PEG chain length. This research therefore adds a new element to the methods available to control polymersome stability and gives new insights into the stabilizing power of PEG chains on the polymersome surface. An overview of the procedure followed is depicted schematically in Fig. 1.
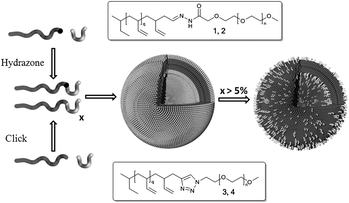 | ||
| Fig. 1 Formation of an inert and an acid labile amphiphilic block copolymerviahydrazone formation (1 and 2) and click chemistry (3 and 4), respectively. These polymers readily form polymersomes. Lowering the pH will hydrolyse the hydrazones, shedding the poly(ethylene glycol) shell. This will either result in fully disrupting the vesicle or reducing the degree of polymersome PEGylation. | ||
The different types of block copolymers as depicted in Fig. 1 were synthesized starting with the preparation of polybutadiene by means of anionic polymerisation. The living polymer was either terminated with a protected aldehyde or alkyne.29 PEG with a molecular weight of 1000 and 2000 g mol−1 was either functionalized with an azide and coupled to polybutadiene by means of click chemistry30 to obtain the inert block copolymers 3 and 4, or modified with a hydrazine to obtain 1 and 2 by hydrazone formation. Table 1 summarizes the properties of polymers 1–4. In Fig. 2 the GPC results of the coupling viahydrazone formation are depicted. This reaction proceeded readily in dichloromethane without the addition of a catalyst. The desired block copolymers were in all cases obtained with a polydispersity well below 1.20 as determined by SEC.
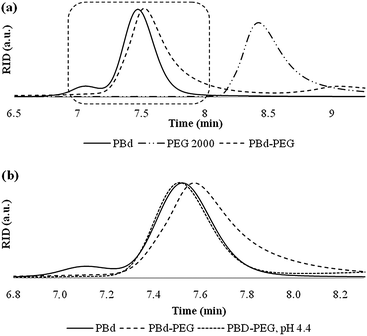 | ||
| Fig. 2 GPC traces (a) of coupling of polybutadiene and PEG viahydrazone formation and (b) after incubation of 2 at pH 4.4 the original polybutadiene chain (A) is obtained back. | ||
To determine the pH sensitivity it was investigated whether the obtained hydrazone-functional polymers could be fully cleaved at low pH, or whether an equilibrium between polymer 1/2 and their cleaved products was obtained. For this purpose polymers 1 and 2 were assembled into polymersomes, after which the pH was lowered to 4.4. Subsequent TLC analysis showed no traces of the block copolymer, only the free PEG and polybutadiene blocks were observed. These results were confirmed by GPC analysis. As can be observed from Fig. 2 no significant amount of block copolymer was present after one hour incubation at pH 4.4.
The pH dependent stability of polymersomes, assembled fully from cleavable polymers 1 or 2, was evaluated next. A standard solution of extruded polymersomes with an average diameter of around 200 nm was prepared. The pH of this solution was 7.5, so no significant hydrolysis could take place in the time frame of sample preparation (see Fig. 3a). After injection of a polymersome sample in buffers ranging in pH, the size distribution was monitored over time by dynamic light scattering (DLS). This method was useful to study destabilization of the polymersomes, since we envisioned that whenever enough PEG was cleaved off, the bare polybutadiene would start to aggregate and form larger, more polydisperse aggregates which eventually would phase separate with water. The DLS results as depicted in Fig. 3 show two trends. First of all Fig. 3a shows how below pH 5.4 polymersomes assembled from 1 lost their stability within one hour and started to aggregate, whereas at physiological pH (7.4) the vesicles were stable in solution for more than three days. Secondly, the DLS curves plotted in Fig. 3b show a marked difference in destabilization rate between pBd–PEG 1 and 2 while incubating in the same buffer, pH 6.4. Whereas polymersomes composed of 1, with the shorter PEG block of 1000 g mol−1, started to aggregate after three hours, the time needed to induce aggregation of polymersomes composed of 2, (with a PEG length of 2000 g mol−1) was more than doubled. These results suggest that the time dependent stability of these polymeric vesicles can be tuned by adjusting the block copolymer composition. Furthermore, these polymersomes are stable at physiological pH but readily aggregate under slightly acidic conditions. What is not clear from these results is whether the gain in stability is a result of slower hydrolysis or better shielding, if longer peripheral PEG chains are applied.
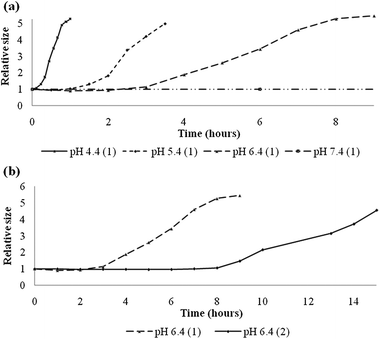 | ||
| Fig. 3 (a) Relative size distribution of polymersomes composed of 1 as a function of time in hours upon incubation in buffers of different pH. (b) Relative size distribution of polymersomes composed of 1 and 2 at pH 6.4 as a function of time in hours. | ||
Via this experiment it is also not possible to determine the composition of the vesicles at the point where aggregation starts to occur. It is actually the bending point in the graph which is interesting because this reflects the composition in which there is just enough PEGylation left to stabilize the polybutadiene sphere. To estimate the polymersome composition at the bending points we decided to prepare mixed polymersomes composed of an inert amphiphilic block copolymer (3 or 4) and a pH sensitive one (1 and 2). Furthermore, in the previous section we showed that at pH 4.4 block copolymers 1 and 2 are fully cleaved and there is no sign of an equilibrium. We therefore exposed the mixed polymersomes to a medium with a pH of 4.4. At this point all cleavable PEG chains were removed, leaving a polybutadiene bilayer displaying only the non-cleavable PEG chains as shown in Fig. 1. After three days all samples were re-measured with DLS to check whether the particle size remained the same or aggregation had occurred. These measurements were performed for both the PEG1000 and PEG2000 analogues for which the results are depicted in Table 2. All samples were stable even after cleaving off 90 percent of the PEG chains. The first sample to become unstable consisted of polymersomes that had a ratio of 95 percent of 1 to 5 percent of 3. This actually means that a little more than 5 percent PEGylation with PEG1000 is enough to stabilize the polybutadiene sphere. Upon doubling the chain length to PEG2000 we were able to remove more than 95 percent of the periphery without disrupting the system. This means that when longer PEG chains are employed even less than 5 percent PEGylation will efficiently stabilize these polymersomes.
| Time/h | 0% | 85% | 90% | 95% | 100% | |
|---|---|---|---|---|---|---|
| 1 | 0 | 227 | 230 | 239 | 226 | 208 |
| 72 | 234 | 220 | 247 | 2000+ | 2000+ | |
| 2 | 0 | 208 | 212 | 251 | 223 | 201 |
| 72 | 208 | 215 | 244 | 242 | 2000+ |
Although this low amount of PEGylation needed to stabilize polymersomes is at first instance somewhat surprising, it can easily be rationalized by considering PEGylated liposomal systems. In liposomal formulations it is common to introduce five to ten percent PEGylation to induce longer blood circulation times by blocking all interactions with the environment. Furthermore, it has been shown for cationic lipid vesicles that ten percent of PEGylation is enough to block all cell interactions, basically shielding a positively charged bilayer from its surrounding.31 Finally, a theoretical study by Smart et al. showed how only a very limited amount of PEG chains can cover a surface by either adopting a mushroom or a fully stretched shape depending on the PEG density.6
In conclusion, we have constructed polymersomes composed of a mixture of stable and pH sensitive, cleavable polybutadiene–PEG amphiphilic block copolymers. With these mixed polymersomes we were able to determine the minimum amount of poly(ethylene glycol) needed for stabilization. At physiological pH these polymersomes retained their colloidal stability for at least three days. Under slightly acidic conditions polymersome stability was only lost when the degree of PEGylation was lowered to 5%. This percentage could be even further decreased by doubling the poly(ethylene glycol) molecular weight.
This study/work was performed within the framework of the Dutch Top Institute Pharma project # T5-105. The authors would like to thank Prof. Dr Dick Hoekstra for the fruitful discussions.
Notes and references
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef.
- P. P. Ghoroghchian, P. R. Frail, K. Susumu, D. Blessington, A. K. Brannan, F. S. Bates, B. Chance, D. A. Hammer and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2922–2927 CrossRef CAS.
- J. Z. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- F. H. Meng, Z. Y. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS.
- T. P. Smart, O. O. Mykhaylyk, A. J. Ryan and G. Battaglia, Soft Matter, 2009, 5, 3607–3610 RSC.
- J.-M. Schumers, J.-F. Gohy and C.-A. Fustin, Polym. Chem., 2010, 1, 161–163 RSC.
- J. S. Katz, S. Zhuong, B. G. Ricart, D. J. Pochan, D. A. Hammer and J. A. Burdick, J. Am. Chem. Soc., 2010, 132, 3654–3655 CrossRef CAS.
- N. Ma, Y. Li, H. P. Xu, Z. Q. Wang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 442–443 CrossRef CAS.
- H. L. Sun, B. N. Guo, R. Cheng, F. H. Meng, H. Y. Liu and Z. Y. Zhong, Biomaterials, 2009, 30, 6358–6366 CrossRef CAS.
- J. H. Ryu, S. Park, B. Kim, A. Klaikherd, T. P. Russell and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 9870–9871 CrossRef CAS.
- A. Klaikherd, S. Ghosh and S. Thayumanavan, Macromolecules, 2007, 40, 8518–8520 CrossRef CAS.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS.
- Y. T. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726–2728 CrossRef CAS.
- A. Napoli, M. J. Boerakker, N. Tirelli, R. J. M. Nolte, N. A. J. M. Sommerdijk and J. A. Hubbell, Langmuir, 2004, 20, 3487–3491 CrossRef CAS.
- S. Yurt, U. K. Anyanwu, J. R. Scheintaub, E. B. Coughlin and D. Venkataraman, Macromolecules, 2006, 39, 1670–1672 CrossRef CAS.
- S. Lin, F. S. Du, Y. Wang, S. P. Ji, D. H. Liang, L. Yu and Z. C. Li, Biomacromolecules, 2008, 9, 109–115 CrossRef CAS.
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelinkova and B. Rihova, J. Controlled Release, 2003, 87, 33–47 CrossRef CAS.
- W. Chen, F. H. Meng, R. Cheng and Z. Y. Zhong, J. Controlled Release, 2010, 142, 40–46 CrossRef CAS.
- R. Jain, S. M. Standley and J. M. J. Frechet, Macromolecules, 2007, 40, 452–457 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS.
- F. H. Meng, C. Hiemstra, G. H. M. Engbers and J. Feijen, Macromolecules, 2003, 36, 3004–3006 CrossRef CAS.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS.
- T. P. King, S. W. Zhao and T. Lam, Biochemistry, 1986, 25, 5774–5779 CrossRef CAS.
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 832–837 CrossRef CAS.
- R. M. Sawant, J. P. Hurley, S. Salmaso, A. Kale, E. Tolcheva, T. S. Levchenko and V. P. Torchilin, Bioconjugate Chem., 2006, 17, 943–949 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS.
- L. He, Y. Jiang, C. Tu, G. Li, B. Zhu, C. Jin, Q. Zhu, D. Yan and X. Zhu, Chem. Commun., 2010, 46, 7569–7571 RSC.
- A. Hirao and M. Hayashi, Acta Polym., 1999, 50, 219–231 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57–59 RSC.
- F. X. Shi, L. Wasungu, A. Nomden, M. C. A. Stuart, E. Polushkin, J. B. F. N. Engberts and D. Hoekstra, Biochem. J., 2002, 366, 333–341 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation. See DOI: 10.1039/c0py00316f |
| This journal is © The Royal Society of Chemistry 2011 |