Controlled/living radical polymerization in nanoreactors: compartmentalization effects
Per B.
Zetterlund
*
Centre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: p.zetterlund@unsw.edu.au; Fax: +61 2 9385 6250; Tel: +61 2 9385 4331
First published on 4th October 2010
Abstract
Compartmentalization in nanoreactors, i.e. the confinement of reactants to monomer droplets or polymer particles with diameters in the approximate range 20–200 nm, may have a marked beneficial effect on the progression of a controlled/living radical polymerization based on the persistent radical effect such as nitroxide-mediated radical polymerization and atom transfer radical polymerization. Compartmentalization effects comprise the confined space effect, which acts to improve the control over the molecular weight distribution (narrower) and the segregation effect which results in increased livingness (end-functionality). Exploitation of nanoreactors thus offers novel means of improving the performance of controlled/living radical polymerizations.
 Per B. Zetterlund | Per B. Zetterlund graduated from The Royal Institute of Technology (Sweden) in 1994 and obtained his Ph.D. at Leeds University (UK) in 1998. He carried out postdoctoral research at Griffith University (Australia), and was appointed Assistant Professor at Osaka City University (Japan) in 1999. In 2003, he moved to Kobe University (Japan) where he was promoted to Associate Professor in 2005. A/ Prof Zetterlund joined CAMD at The University of New South Wales (Australia) in 2009. Current research focuses on controlled/living radical polymerization in dispersed systems for synthesis of polymeric nanoparticles. A/ Prof Zetterlund has published 92 peer-reviewed papers and 2 book chapters. |
Introduction and scope
Compartmentalization refers to the physical confinement of reactants within discrete confined spaces, so-called nanoreactors. The term nanoreactor simply refers to a nanoscale chemical reactor. In the field of radical polymerization, by nanoreactors one normally refers to monomer droplets, monomer-swollen micelles or polymer particles with diameters lower than approximately 200 nm (depending on the particular system) during polymerization in dispersed systems (emulsion, miniemulsion, etc.). Radical polymerization can be performed in various dispersed systems, e.g. emulsion,1 miniemulsion,2–4 microemulsion,5,6 precipitation, dispersion7,8 and suspension polymerization systems, where the continuous phase is usually, but not always,9,10 water.Compartmentalization effects in radical polymerization become significant if some fraction of droplets/particles contain a sufficiently low number of the relevant reactant(s). This occurs if the polymerization recipe is such that a very low concentration of propagating radicals is generated, and/or if the particles where polymerization occurs are sufficiently small. The concept of compartmentalization and the use of nanoreactors are also being exploited in other areas of chemistry.11
The influence of compartmentalization on the kinetics of conventional non-living emulsion polymerization systems is relatively well-understood.1 The polymerization rate (Rp) and the obtained polymer molecular weight in an emulsion polymerization are generally higher than in the corresponding homogeneous (bulk/solution) polymerization as a consequence of compartmentalization of propagating radicals leading to reduced bimolecular termination rates. The last two decades have seen the development of a range of techniques of controlled/living radical polymerization (CLRP),12 which enable precise polymer synthesis, good control over molecular weight distributions (MWDs) and make various complex polymer architectures accessible. Moreover, CLRP in dispersed systems enables synthesis of nanoparticles comprising polymer of well-defined microstructure as well as industrial application of CLRP.13,14 By careful consideration of the experimental conditions, it is possible to exploit compartmentalization effects in CLRP to improve both the livingness (end-functionality) and the level of control over the MWD (narrower MWD).
The vast majority of the CLRP systems developed to date proceed via one of the two basic mechanisms: (i) the persistent radical effect (PRE)15–17 and (ii) degenerative transfer.17 Nitroxide-mediated radical polymerization (NMP, Scheme 1)18,19 and atom transfer radical polymerization (ATRP, Scheme 2)12,20,21 are based on the PRE, whereas reversible addition-fragmentation chain transfer (RAFT)22,23 polymerization proceeds via degenerative transfer. The present review is concerned with the effects of compartmentalization in CLRP systems that operate via the PRE, and is a comprehensive account of all relevant experimental and theoretical work published to date (July of 2010) in this field.
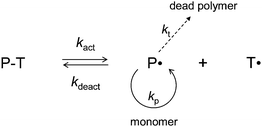 | ||
| Scheme 1 Nitroxide-mediated radical polymerization (NMP). | ||
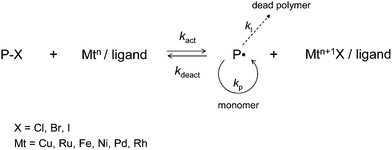 | ||
| Scheme 2 Atom transfer radical polymerization (ATRP). | ||
Fundamental concepts of compartmentalization in CLRP
All CLRP systems developed to date are based on propagating radicals being reversibly deactivated, i.e. alternating between active and dormant states.17 In NMP, the dormant state is a polymeric alkoxyamine, in ATRP it is a polymeric alkyl halide, and in RAFT it is a polymer chain with a RAFT end-group. The term “livingness” refers to the number fraction of polymer chains that are dormant and can be chain extended. The term “control” refers to the number-average molecular weight (Mn) increasing linearly with conversion and Mw/Mn decreasing with increasing conversion,24i.e. good control in practice means a narrow MWD.Compartmentalization effects in CLRP can be considerably more complex than in conventional non-living radical polymerization systems because in addition to the propagating radicals, the deactivator species (nitroxide in NMP, Cu(II) complex in ATRP) may also be compartmentalized. In both NMP25–36 and ATRP31,37–41 (systems based on the PRE), the deactivation step can relatively easily be influenced by compartmentalization because of the low concentration of deactivator (10−5 to 10−3 M). In degenerative transfer CLRP systems (e.g. RAFT), the deactivation step (i.e. reaction between propagating radical and RAFT end group) is not influenced by compartmentalization under normal conditions because the concentration of RAFT end groups is too high.42–45
There are two fundamental types of compartmentalization effects: the segregation effect and the confined space effect (Fig. 1).25 The segregation effect refers to two species located in separate particles being unable to react, and is the reason that both Rp and the molecular weight are normally higher in an emulsion polymerization than in the corresponding homogeneous system. The rate of bimolecular termination between propagating radicals is reduced due to propagating radicals being segregated by confinement to individual particles.
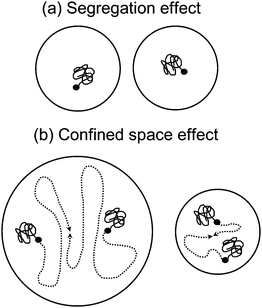 | ||
| Fig. 1 Schematic illustrations of (a) the segregation effect and (b) the confined space effect in a compartmentalized reaction system. Reprinted with permission from ref. 13. Copyright 2008 American Chemical Society. | ||
The confined space effect refers to two species located in the same particle reacting at a higher rate in a small particle than in a large particle. The rate of reaction between two species in the same particle increases with decreasing particle size, as given by the pseudo first-order rate coefficient k/NAvp (k is the rate coefficient for a bimolecular reaction in M−1 s−1, NA is Avogadro's number, and vp is the particle volume). The confined space effect is perhaps most readily illustrated when comparing the deactivation reaction in NMP between a propagating radical (P˙) and nitroxide (T˙) in a compartmentalized system with the corresponding homogeneous system as illustrated in Fig. 2.34 When the organic phase is divided into discrete particles, there will be some particles that do not contain any T˙ if the overall concentration of T˙ is sufficiently low and/or the particles are sufficiently small. Under such circumstances, as shown in Fig. 2a, the concentration of T˙ in the particles that do contain T is higher than in the homogeneous system (because the volume in particles with no T˙ is “excluded”). Consequently, the rate of deactivation per P˙, given by kdeact[T˙] (s−1), is higher in the dispersed system. In the case of propagation, the concentration of monomer (M) is so high that there are no particles with no M, and thus there is no confined space effect on propagation (in a real system, the number of monomer units per particle is of course much higher than in the schematic illustration). Bimolecular reactions are not influenced by the confined space effect if the concentration of one of the species is too high. This is why there is no confined space effect in RAFT systems—the concentration of RAFT end groups is usually of the order of 10−2 M, and thus all particles contain RAFT end groups. The same applies to the bimolecular activation reaction in ATRP (alkyl bromide reacting with Cu(I) complex)—the concentration of the Cu(I) complex is too high (in NMP, activation is a first order reaction, and thus unaffected by compartmentalization). This criterion for a confined space effect to be operative is expressed mathematically in eqn (1):30
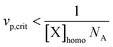 | (1) |
![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) X = [X]NAvp,crit = 1, where X denotes the average number of X per particle. However, even in situations where X = 1 or even if X is somewhat greater than 1, there will be a statistical distribution of X over the total number of particles, and thus some particles would contain no X, whereas others would contain two or three or more X. Consequently, the confined space effect may operate at vp > vp,crit.
X = [X]NAvp,crit = 1, where X denotes the average number of X per particle. However, even in situations where X = 1 or even if X is somewhat greater than 1, there will be a statistical distribution of X over the total number of particles, and thus some particles would contain no X, whereas others would contain two or three or more X. Consequently, the confined space effect may operate at vp > vp,crit.
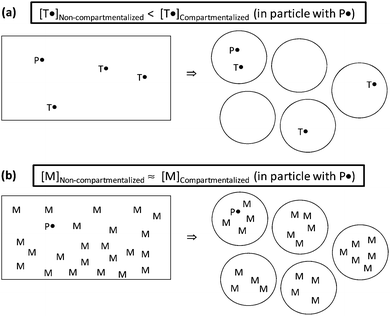 | ||
| Fig. 2 Schematic illustrations of the confined space effect. (a) The confined space effect can be operative on the deactivation reaction. (b) The confined space effect is not operative on the propagation reaction. Reprinted with permission from ref. 34. Copyright 2010 Wiley-VCH Verlag GmbH & Co. | ||
The confined space effect in CLRP was first reported by Zetterlund and Okubo in 2006 based on modeling and simulations.25 In 2007, Tobita verified the findings by Zetterlund and Okubo using a different modeling approach, but referred to the Confined Space Effect as the Single Molecule Concentration (SMC) Effect.30 Both terms refer to the same phenomenon.
One might wonder, why is there no confined space effect operating on the bimolecular termination reaction in CLRP, despite the fact that the number of propagating radicals per particle p˙ ≪ 1? The reason is that propagating radicals (P˙) are not generated in pairs in the same particle, but appear one by one in separate particles. An activation event in NMP, for example, generates a P˙ and a nitroxide molecule (T˙) in the same particle. If P˙ and T˙ were generated in separate particles, there would be no confined space effect on deactivation, but instead a segregation effect.
The confined space effect is not operative on the deactivation reaction in NMP and ATRP (or any CLRP system based on the PRE) if the deactivator is able to undergo transport between particles (phase transfer) sufficiently rapidly.33,46,47 Transport between particles refers to exit from one particle, subsequent diffusion through the continuous phase followed by entry into another particle. Propagating radicals are too hydrophobic to undergo exit and are thus always confined to a given particle, with the possible exception of very short radicals originating from initiator decomposition or chain transfer to monomer.28
The overall effects of compartmentalization on CLRP are quite complex and very dependent on the particular system and the conditions. In the simplest of terms, the segregation effect on bimolecular termination results in an increase in livingness (higher end-functionality), whereas the confined space effect on deactivation may result in an increase in the level of control over the MWD (narrower MWD). However, under some circumstances, compartmentalization may also result in broadening of the MWD.
Theoretical investigations of compartmentalization in CLRP
Modified Smith–Ewart equations
The Smith–Ewart equations48 for analysis of conventional non-living emulsion polymerization systems were developed over half a century ago. They are balance equations that keep track of the number of propagating radicals per particle and form the basis of emulsion polymerization theory.1 Modified two-dimensional Smith–Ewart equations, originally derived by Butte et al.,49 have been employed to investigate compartmentalization effects in NMP25,26,32–36 and ATRP37,39,40 in dispersed systems by Zetterlund and co-workers, and recently also by Thompson and Cunningham (for ATRP).41 The modified two-dimensional Smith–Ewart equations, given below for the case of NMP, account for compartmentalization of both propagating radicals and deactivator species: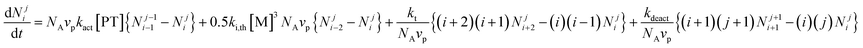 | (2) |
Monte Carlo simulations
Tobita and Yanase have studied compartmentalization effects in NMP29–31 and ATRP31 systems using Monte Carlo simulations by extending an earlier technique52 developed for conventional non-living emulsion polymerization to apply to CLRP systems. The progression of the polymerization is simulated for one single particle a multitude of times, thus providing information on how an entire dispersed systems (comprising numerous particles) behaves. All possible reactions are considered in terms of probabilities, and each kinetic event (reaction) alters the probabilities for the next kinetic event.Both modeling approaches described above (modified Smith–Ewart equations and Monte Carlo modeling) are based on models that correspond to an ideal miniemulsion polymerization,2 where the system initially comprises monomer droplets that are subsequently converted to polymer particles. The total number of monomer droplets (polymer particles) remains constant, i.e. there is no secondary nucleation or Ostwald ripening.53 However, the results may also be invoked, with caution, to understand the kinetics of CLRP in other dispersed systems, e.g. emulsion polymerization. Phase transfer events (most notably exit and subsequent entry of deactivator, i.e. nitroxide or Cu(II) complex) are not accounted for in the models. Depending on the particular system, it cannot be excluded that such events may to some extent influence a real system, as dealt with in detail later in this review.
Theory: compartmentalization in NMP
This section is concerned with the understanding of compartmentalization effects in NMP that has emerged based on modeling and simulations using modified Smith–Ewart equations and Monte Carlo methods. To date, modeling and simulations based on modified Smith–Ewart equations have been performed for St with the nitroxides TEMPO,25,26,36 SG1,33 TIPNO,32,36 and for butyl acrylate/TEMPO35 (Scheme 3). Monte Carlo simulations have been carried out for St/TEMPO.29,30 | ||
| Scheme 3 Nitroxides used in NMP. | ||
Compartmentalization effects in NMP are best illustrated by considering specific examples. In what follows, compartmentalization effects for NMP of styrene based on the nitroxides TEMPO and TIPNO are described by examination of the influence of particle size on Rp, livingness and control.
Polymerization rate
Fig. 3a shows simulated conversion–time data for TEMPO-mediated radical polymerization of styrene in bulk and a dispersed system.32 For particle diameters (d) > 70 nm, Rp(bulk) ≈ Rp(disp. system), but as d is reduced below approximately 70 nm, Rp decreases dramatically. To understand the effect of particle size on Rp, it can be more instructive to instead plot log [P˙] vs. log d at a fixed monomer conversion (Rp is proportional to [P˙] viaRp = kp[P˙][M] because propagation is not affected by compartmentalization). Such a plot is displayed in Fig. 4 at 1% monomer conversion, revealing how for TEMPO, [P˙] decreases with decreasing particle size. Close examination of the data reveals that there is a particle size region (d ≈ 55–95 nm) where [P˙] is in fact slightly higher than in bulk. The results for the TIPNO/St system are quite different (Fig. 3b and 4). Rp is similar to the bulk system for d-values somewhere in between 10 and 15 nm. For larger particles than this, Rp is greatly increased relative to bulk, whereas the opposite is true for d < 10–15 nm.![Simulated conversion vs. time for different particle diameters (in nm as indicated) for TEMPO- (a) and TIPNO- (b) mediated radical polymerization of styrene in dispersed system at 125 °C (spontaneous initiation of styrene included, [PT]0 = 0.02 M). The dotted line denotes simulated bulk conditions. Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co.](/image/article/2011/PY/c0py00247j/c0py00247j-f3.gif) | ||
| Fig. 3 Simulated conversion vs. time for different particle diameters (in nm as indicated) for TEMPO- (a) and TIPNO- (b) mediated radical polymerization of styrene in dispersed system at 125 °C (spontaneous initiation of styrene included, [PT]0 = 0.02 M). The dotted line denotes simulated bulk conditions. Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co. | ||
![Simulated propagating radical concentrations vs. particle diameter for TIPNO (●) and TEMPO (○) mediated radical polymerization of styrene in dispersed system at different particle diameters at 125 °C (spontaneous initiation of styrene included, [PT]0 = 0.02 M) at 1% styrene conversion. The dotted lines denote the simulated propagating radical concentrations under bulk conditions. Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co.](/image/article/2011/PY/c0py00247j/c0py00247j-f4.gif) | ||
| Fig. 4 Simulated propagating radical concentrations vs. particle diameter for TIPNO (●) and TEMPO (○) mediated radical polymerization of styrene in dispersed system at different particle diameters at 125 °C (spontaneous initiation of styrene included, [PT]0 = 0.02 M) at 1% styrene conversion. The dotted lines denote the simulated propagating radical concentrations under bulk conditions. Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co. | ||
The effects of compartmentalization on deactivation and termination can be elucidated by an imaginary experiment, where the dispersed phase is instantaneously converted into a continuous bulk phase, and the compartmentalized deactivation and termination rates (Rc) are compared with the corresponding non-compartmentalized (Rnc) rates according to eqn (3)–(6).25 The non-compartmentalized system is not a real bulk system, but a hypothetical bulk system where the concentrations of P˙ and T˙ correspond to the overall instantaneous organic phase concentrations in a compartmentalized system.
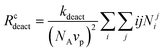 | (3) |
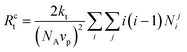 | (4) |
| Rncdeact = kdeact [P˙] [T˙] | (5) |
| Rnct = 2kt[P˙]2 | (6) |
The results, in the form of plots of log(Rc/Rnc) vs. log d, are shown for deactivation and termination in Fig. 5. For sufficiently large particles, log(Rc/Rnc) = 0, indicating that there are no compartmentalization effects on neither deactivation nor termination. For both nitroxides, log(Rc/Rnc) for deactivation increases markedly with decreasing particle size due to the confined space effect. In the case of styrene, spontaneous radical generation occurs from the monomer, resulting in radicals generated in pairs in the same particle.50,51 The confined space effect operates on these radicals, thus causing high rates of geminate termination in small particles. This is the reason that log(Rc/Rnc) for termination in fact increases with decreasing particle size for both nitroxides at sufficiently small particles. In the absence of spontaneous initiation (triangles in Fig. 5), the termination rate is reduced relative to the homogeneous system by segregation also for very small particles.
![Ratios of “compartmentalized” (Rc) and “non-compartmentalized” (Rnc) deactivation (a) and termination (b) rates for TIPNO (●) and TEMPO (○) mediated radical polymerization of styrene in dispersed system at different particle diameters (d) in the presence of thermal initiation at 125 °C ([PT]0 = 0.02 M) at 1% styrene conversion. Data are also shown for the hypothetical cases in the absence of spontaneous initiation of styrene (ki,th = 0) for TIPNO (▲) and TEMPO (△). Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co.](/image/article/2011/PY/c0py00247j/c0py00247j-f5.gif) | ||
| Fig. 5 Ratios of “compartmentalized” (Rc) and “non-compartmentalized” (Rnc) deactivation (a) and termination (b) rates for TIPNO (●) and TEMPO (○) mediated radical polymerization of styrene in dispersed system at different particle diameters (d) in the presence of thermal initiation at 125 °C ([PT]0 = 0.02 M) at 1% styrene conversion. Data are also shown for the hypothetical cases in the absence of spontaneous initiation of styrene (ki,th = 0) for TIPNO (▲) and TEMPO (△). Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co. | ||
The dramatic reduction in Rp for very small particles is thus a result of the confined space effect on deactivation. Activation generates P˙ and T˙ in the same particle, and the smaller the particle, the more rapid is the deactivation reaction between the same P˙ and T˙ (prior to which monomer addition to P˙ may or may not occur). For sufficiently small particles, the slope of log [P˙] vs. log d is equal to 3, which is a consequence of the vast majority of particles containing no P˙ and no T˙, and the pseudo-first-order deactivation rate coefficient (kdeact/NAvp) increasing in proportion to d−3 with decreasing particle size.25 For the St/TEMPO/125 °C system, at 10% St conversion, P˙ increases from 4.2 × 10−8 to 1.7 × 10−3 and T˙ from 3.4 × 10−5 to 2.9 as the particle diameter is increased from 10 to 70 nm.25
Based on the data displayed in Fig. 5, the interplay between three factors govern whether Rp is higher or lower than in the corresponding bulk system: (i) the confined space effect on deactivation, (ii) the confined space effect on geminate termination of spontaneously generated radicals from styrene, and (iii) the segregation effect on termination. (i) and (ii) lead to a reduction in Rp, whereas (iii) leads to an increase in Rp. It follows that the maximum in Rp can be rationalized based on the segregation effect on termination dominating over the confined space effect in that particle size region.
However, an additional factor may also be at play. As proposed by Tobita,30 the so-called fluctuation effect refers to how fluctuation in the number of T˙ between different particles can lead to a higher overall [P˙] (and thus Rp) in the total organic phase than what would be the case if all particles contained the same number of T˙. For a given concentration of living (dormant) chains, the value of Rp is proportional to the average number of monomer units added during an activation–deactivation cycle (N), which is given by:30
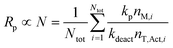 | (7) |
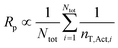 | (8) |
The fluctuation effect is illustrated by comparing Rp as given by eqn (8) for a case where T˙ species are unevenly distributed with the case where all particles contain the same number of T˙ as given by T,Act,i (the average number of T˙), according to eqn (9):
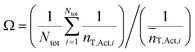 | (9) |
There is a rate enhancement due to the fluctuation effect if Ω > 1. As illustrated in the original paper by Tobita,30 if we consider two systems with T,Act,i = 3, where all particles contain 3 T˙ in system 1, and 50% of particles contain 1 T˙ and 50% contain 5 T˙ in system 2, eqn (9) then tells us that Rp will be 80% higher in system 2 (Ω = 1.8). The fluctuation effect is significant if the number of T˙ per particle is less than approximately 10 (as is the case when the confined space effect operates), and the maximum effect on [P˙] and Rp is an increase of approximately a factor of two compared to a system where all particles contain the same number of T˙.30
Monte Carlo simulations have indicated that in some cases, an increase in Rp may be caused entirely by the fluctuation effect, with negligible contribution by the segregation effect (the latter referred to as the “isolation effect” in Tobita's papers).30 The segregation effect leads to a reduction in [T˙] (less termination means that less T˙ is “released”), and Monte Carlo simulations showed that under certain conditions [T˙] in “active” particles (particles containing P˙) is very similar to [T˙] in the corresponding bulk (homogeneous) system, yet Rp(dispersed system) > Rp(bulk), consistent with the fluctuation effect causing the enhanced Rp. Fluctuations in the number of T˙ between particles are accounted for in simulations employing the modified Smith–Ewart equations. Fluctuation in the number of T˙ between particles affects the way in which both the overall deactivation and termination rates are influenced by compartmentalization, as computed with the Smith–Ewart equations. However, in the Smith–Ewart simulations for the system St/TEMPO/125 °C,25 which correspond to very similar conditions and rate coefficients as the system simulated using the Monte Carlo technique,30 it can be readily shown that [T˙]particle < [T˙]bulk (for “active” particles), which is consistent with the segregation effect being operative. The same conclusion is reached by applying the same analysis to the results of the Smith–Ewart simulations for the system St/TIPNO/125 °C.32 Moreover, the extent of the maximum in Rp relative to bulk for the system St/TIPNO/125 °C is a factor of 13.8, much more than what can be accounted for based on the fluctuation effect.
Livingness
The term “livingness” refers to the extent of end-functionality, i.e. in the case of NMP, the number fraction of chains that possess an alkoxyamine end-functionality and are thus able to undergo further chain extension. Fig. 6 shows the livingness vs. conversion for different particle sizes for the systems St/TEMPO/125 °C25 and St/TIPNO/125 °C,32 expressed as the number fraction of chains with alkoxyamine end-functionality relative to the initial amount (the differences between the two systems are discussed below). The livingness in the compartmentalized systems is greater than in the corresponding bulk systems, and the livingness increases with decreasing particle size (data for the latter not shown). This is a consequence of the segregation effect leading to reduced levels of termination between propagating radicals. Compartmentalization effects in the form of the segregation effect always lead to higher livingness. The influence of the fluctuation effect on the degree of livingness has to date not been elucidated, but is most likely marginal under the typical conditions described in this review.![Simulated values of fraction of alkoxyamine PT (rel. to initial amount) as function of conversion for TIPNO (thick lines) and TEMPO (thin lines) mediated radical polymerization of styrene in dispersed system in the presence of spontaneous initiation of styrene at 125 °C ([PT]0 = 0.02 M). Full lines: bulk. Broken lines: dispersed system with particle diameters (d) 40 nm. Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co.](/image/article/2011/PY/c0py00247j/c0py00247j-f6.gif) | ||
| Fig. 6 Simulated values of fraction of alkoxyamine PT (rel. to initial amount) as function of conversion for TIPNO (thick lines) and TEMPO (thin lines) mediated radical polymerization of styrene in dispersed system in the presence of spontaneous initiation of styrene at 125 °C ([PT]0 = 0.02 M). Full lines: bulk. Broken lines: dispersed system with particle diameters (d) 40 nm. Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co. | ||
Control over MWD
The control over the MWD refers to the narrowness of the MWDs—the narrower the MWD, the better is the control (lower polydispersity, Mw/Mn). In the case of ideal CLRP (absence of termination, transfer and other side reactions), the polydispersity is dictated by the number of activation–deactivation cycles that occur as polymer chains grow to their final molecular weight.17 The higher the number of cycles, the narrower is the MWD and the lower is Mw/Mn. Consequently, for a given molecular weight, the number of activation–deactivation cycles is inversely proportional to the number of monomer units added per activation–deactivation cycle for an individual chain (N). Full MWDs have not been computed based on the Smith–Ewart simulations. Instead, the level of control has been assessed via the quantity N as given by eqn (10):25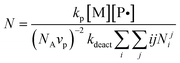 | (10) |
It is noted that the eqn for N accounts for fluctuation effects, i.e.N is not calculated from P˙ and T˙. Fig. 7 shows Nvs. conversion for different particle sizes for the systems St/TEMPO/125 °C25 and St/TIPNO/125 °C,32 revealing that for both systems, the control is superior to in bulk (i.e.N is lower) if the particles are sufficiently small. The narrowing of the MWD for small particles is a result of the confined space effect, which results in an increase in the rate of deactivation relative to the rate of propagation. However, as illustrated for the TIPNO system, if the particle diameter exceeds some critical value, the level of control is lower than in bulk (i.e. higher N). This situation corresponds to when there is an increase in Rp relative to the corresponding bulk system (Fig. 3 and 4). Under such conditions, the segregation effect leads to a reduction in the extent of termination, which in turn causes a low [T˙], and thus a low deactivation rate. In other words, the segregation effect (causing low termination rate) dominates over the confined space effect (causing high deactivation rate), and the end result is an increase in both N and Rp.
![Simulated numbers of propagation events per activation–deactivation cycle for an individual chain (N) as function of conversion for TIPNO (thick lines) and TEMPO (thin lines) mediated radical polymerization of styrene in dispersed system in the presence of spontaneous initiation of styrene at 125 °C ([PT]0 = 0.02 M). Full lines: bulk. Broken lines: dispersed system with particle diameters (d) as indicated. Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co.](/image/article/2011/PY/c0py00247j/c0py00247j-f7.gif) | ||
| Fig. 7 Simulated numbers of propagation events per activation–deactivation cycle for an individual chain (N) as function of conversion for TIPNO (thick lines) and TEMPO (thin lines) mediated radical polymerization of styrene in dispersed system in the presence of spontaneous initiation of styrene at 125 °C ([PT]0 = 0.02 M). Full lines: bulk. Broken lines: dispersed system with particle diameters (d) as indicated. Reprinted with permission from ref. 32. Copyright 2009 Wiley-VCH Verlag GmbH & Co. | ||
The fluctuation effect may also contribute towards an increase in N (see eqn (7)). Moreover, if different particles contain different numbers of T˙, Mn will be different in different particles, thus causing MWD broadening.30 Such a case is illustrated by Monte Carlo simulations in Fig. 8, showing how for a particle diameter of 30 nm, the confined space effect dominates giving Mw/Mn lower than in bulk, but for larger particles the fluctuation effect contributes to Mw/Mn being greater than in bulk.
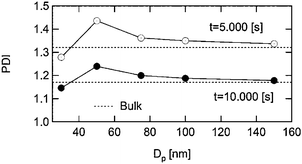 | ||
| Fig. 8 Simulated polydispersity (Mw/Mn) vs. particle diameter for NMP in a dispersed system at two different polymerization times (see original publication for details). The dotted lines show Mw/Mn in the corresponding bulk system. Reprinted with permission from ref. 29. Copyright 2007 Wiley-VCH Verlag GmbH & Co. | ||
Effect of NMP equilibrium constant
Compartmentalization effects in NMP are strongly dependent on the particular NMP system. In short, the lower the NMP equilibrium constant (K = kact/kdeact, Scheme 1), the more likely it is that compartmentalization will influence the polymerization. This is because the lower the value of K, the lower are bothP˙ and T˙ at a given particle size (if P˙ and T˙ are too high, the system is not compartmentalized; the same logic can be applied to the effect of dilution26,34).
The effects of K are clearly illustrated by comparing the systems St/TEMPO/125 °C25 and St/TIPNO/125 °C;32K is 357 times greater for TIPNO/St than TEMPO/St at 125 °C as detailed in Table 1. The main difference between TEMPO and TIPNO is that for TIPNO, a particle size region exists where there is a very significant increase in Rp (Fig. 3 and 4) and loss of control relative to the corresponding bulk system (Fig. 7). Such an increase in Rp and loss of control are also seen for TEMPO in a certain particle size range, but to a much lesser extent.
| St/TEMPO/125 °C | St/TIPNO/125 °C | |
|---|---|---|
| k act/s−1 | 1.60 × 10−3 | 3.20 × 10−3 |
| k deact/M−1 s−1 | 7.60 × 107 | 4.27 × 105 |
| K/M | 2.1 × 10−11 | 7.5 × 10−9 |
The effects of compartmentalization on deactivation are relatively similar for TIPNO and TEMPO, although smaller particles are required for the confined space effect to be operative for TIPNO (Fig. 5). The effect of the value of K on the extent of the segregation effect on termination is much more pronounced (Fig. 5). The segregation effect on propagating radicals is very strong for TIPNO, the termination rate being much lower in the compartmentalized system than in bulk (except for very small particles of d < 10 nm). The relative contribution of radicals from alkoxyamine activation relative to spontaneous initiation of St is larger for TIPNO than TEMPO because K(TIPNO) ≫ K(TEMPO), and thus the contribution of geminate termination of spontaneously generated radicals towards the overall termination rate is smaller for TIPNO than TEMPO. Radicals generated in pairs from spontaneous initiation of styrene terminate rapidly due to the confined space effect in small particles, thus counteracting the observed segregation effect. However, even with the spontaneous initiation rate set to zero, Rct/Rnct (TIPNO) < Rct/Rnct (TEMPO) (triangles in Fig. 5). This difference in the magnitude of the segregation effect in the absence of spontaneous initiation is caused by its intrinsic dependence on the overall propagating radical concentration in the dispersed phase. The fluctuation effect cannot cause Rp enhancement in excess of approximately a factor 2,30 and is therefore not a main factor with regard to the vastly different compartmentalization effects observed between TEMPO and TIPNO.
A pronounced increase in Rp and concomitant decrease in the level of control over the MWD (increase in N) occurs if the segregation effect is sufficiently dominant over the confined space effect. Thus, the fact that the NMP equilibrium is shifted more towards the active state for TIPNO/St than TEMPO/St leads to a particle size region where Rp is much greater than in bulk and where control is essentially lost over the MWD for St/TIPNO. Very small particles are required for control to be obtained in the TIPNO system, i.e. for the confined space effect to be strong enough to overcome the segregation effect. However, under all conditions examined, the livingness is improved by compartmentalization (Fig. 6). The gain in livingness in the TIPNO system is markedly greater than for TEMPO (also in the particle size region where the control is poor for TIPNO) due to the stronger segregation effect for TIPNO.
Theory: compartmentalization in ATRP
This section describes the understanding of compartmentalization effects in ATRP12,20,21 (Scheme 2) that has been gained from modeling and simulations using modified Smith–Ewart equations and Monte Carlo methods. From a kinetic point of view, the mechanism of ATRP is very similar to that of NMP. Both systems are based on an equilibrium between active and dormant chains and the PRE,15,16 the main kinetic difference being that in ATRP, the activation step is bimolecular whereas it is unimolecular in NMP. As stated earlier, the activation step in ATRP is not affected by compartmentalization (because the concentration of the Cu(I) complex is too high), and it follows that the kinetic treatment of NMP applies to ATRP if one simply replaces kact by kact[Cu(I)] in the relevant expression related to the activation step. Consequently, all the features of compartmentalization for NMP described in some detail in the preceding section (e.g. segregation effect on termination, confined space effect on deactivation, effects on Rp, livingness and control) are present also in ATRP systems. To avoid repetition, this section will focus on aspects of compartmentalization that are specific to ATRP and/or have not yet been studied for NMP, and consequently the ATRP section is structured differently from the NMP section above.To date, modeling and simulations based on modified Smith–Ewart equations (eqn (2) adapted to ATRP) have been performed for the systems n-butyl acrylate/CuBr/4,4′-dinonyl-2,2′-dipyridyl (dNbpy2),37 styrene/CuX/dNbpy2 (X = Br or Cl),39,40 and n-butyl methacrylate/CuBr with the tris(2-aminoethyl)amine (TREN) based ligand EHA6TREN41 (Scheme 4).
 | ||
| Scheme 4 ATRP ligands (two different isomers of dNbpy are commonly employed, here denoted dNbpy1 and dNbpy2). | ||
Particle size for compartmentalization
For the system n-butyl acrylate/CuBr/dNbpy2 at 110 °C,37 simulations show that significant compartmentalization effects can be expected for particle diameters less than approximately 70 nm. A small maximum in Rp occurs at 60 nm, where Rp is greater than in bulk by a factor of less than 2. For particle diameters of 50 nm and less, marked retardation occurs due to the confined space effect on deactivation.The effect of the halide, Br or Cl, was investigated for the system styrene/CuX/dNbpy2 (X = Br or Cl) at 70 or 75 °C.39,40 Depending on the halide and the targeted molecular weight (i.e. the concentration of living (dormant) chains), the critical particle diameter below which compartmentalization effects were observed was in the range 30–100 nm. All systems investigated in this study exhibited the characteristic maximum in Rp at a certain particle size, accompanied by some loss of control over the MWD (as assessed viaN) in the particle size range where Rp was greater than in the corresponding bulk system. The magnitude of the increase in Rp at the maximum value varied between factors of 2.1 and 5.3.
The effect of the type of halide is similar to the situation for NMP described above with regard to the effect of nitroxide type. For the system in question, the value of kact/kdeact at 75 °C is approximately 11 times greater for Br than Cl, i.e. the ATRP equilibrium is positioned more towards the active state for Br. As a result, smaller particles are required for compartmentalization effects to be manifested in the Br system. This is illustrated in Fig. 9, which depicts plots of log[P˙] vs. log d for the two systems. Both plots show the characteristic linear region for small particles (where the slope is equal to 3, see NMP section), a maximum at 35 (Br) and 70 (Cl) nm, and log[P˙] approaching the respective bulk values for larger particles. At the maxima, the enhancements in Rp relative to the corresponding bulk systems are factors of 2.6 (Br) and 5.3 (Cl). In both systems, the particle size region with an enhanced Rp also exhibits loss of control (higher N, i.e. broader MWD) than the corresponding bulk system, but improved livingness. The effect of the target molecular weight, i.e. the concentration of living (dormant) chains relative to monomer, is qualitatively the same as the effect of the position of the ATRP equilibrium. The lower the targeted molecular weight, the higher is the concentration of alkyl halide (dormant chains), and consequently the higher are the concentrations of propagating radicals and Cu(II) deactivator, and as a result smaller particles are required for compartmentalization effects to play a role (in complete analogy with the case in NMP26).
![Simulated [P˙] as a function of particle diameter (d) at 1% conversion for ATRP of styrene ([styrene]0 = 8.7 M in organic phase) at 75 °C in a dispersed system as a function of particle diameter (d) and in bulk (dotted lines). (○): [PX]0 = [CuBr/2dNbpy]0 = 43.5 mM; (●) [PX]0 = [CuCl/2dNbpy]0 = 43.5 mM. The dotted lines indicate [P˙] in bulk of the respective bulk systems at 1% conversion. Reprinted with permission from ref. 39. Copyright 2009 American Chemical Society.](/image/article/2011/PY/c0py00247j/c0py00247j-f9.gif) | ||
| Fig. 9 Simulated [P˙] as a function of particle diameter (d) at 1% conversion for ATRP of styrene ([styrene]0 = 8.7 M in organic phase) at 75 °C in a dispersed system as a function of particle diameter (d) and in bulk (dotted lines). (○): [PX]0 = [CuBr/2dNbpy]0 = 43.5 mM; (●) [PX]0 = [CuCl/2dNbpy]0 = 43.5 mM. The dotted lines indicate [P˙] in bulk of the respective bulk systems at 1% conversion. Reprinted with permission from ref. 39. Copyright 2009 American Chemical Society. | ||
Simultaneous increase in Rp and control
In all the theoretical work on compartmentalization in CLRP systems conducted so far, it has not been possible to identify conditions where compartmentalization effects result in a simultaneous increase in Rp and improved control (as assessed by N) and livingness. Simultaneous increases in Rp and livingness occur in the particle size range where the segregation effect dominates over the confined space effect as outlined above for both NMP and ATRP. However, under such conditions, a (partial) loss of control is also observed in the form of a broader MWD and higher Mw/Mn (as assessed viaN). Indeed, based on the fact that the increase in Rp is caused by an increase in the number of monomer units added per activation–deactivation cycle (N), it is tempting to conclude that it is impossible to achieve a simultaneous increase in Rp and the level of control.However, according to the simulations of ATRP in a compartmentalized system employing modified Smith–Ewart equations with n-butyl methacrylate and the highly active catalyst/ligand system CuBr/EHA6TREN reported by Thomson and Cunningham,41 a window exists for this particular system where a simultaneous increase in Rp and the level of control (and livingness) is obtained (Fig. 10). The model employed was an expanded version of that in ref. 37 to also enable computation of Mw/Mn of dormant chains, i.e. the level of control was assessed directly from Mw/Mn, not by examination of N (as in the work by Zetterlund et al.25,26,32–37,39,40). It is important to note that the Mw/Mn computed is that of dormant chains only, i.e. broadening of the MWD due to termination is not accounted for. Fig. 10 shows plots of log chain(chain = P˙/(no. of polymer chains in the system)) and Mw/Mnvs. log d, revealing a particle size region around 45 nm where Rp is greater and Mw/Mn is lower than in bulk. In all previous theoretical work (NMP and ATRP) as well as in this work by Thomson and Cunningham (P˙ is proportional to N), there exists no particle size region where Rp is higher and N is lower than in bulk, because Rp, [P˙] and N are all proportional to one another for a given concentration of dormant chains (i.e. the maxima in Rp and N coincide). In the work by Thomson and Cunningham, the maximum value of Mw/Mn occurs at slightly larger particles than the maximum in N, thus creating a window where an increase in Rp and a decrease in Mw/Mn can be attained. Thus, the question is, is this offset between N and Mw/Mn specific to this particular system or is this a more general phenomenon also for other ATRP and NMP systems?
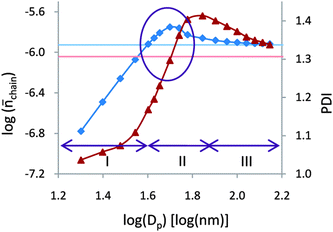 | ||
| Fig. 10 Simulated average number of propagating radicals per particle per chain (chain, squares) and Mw/Mn (PDI, triangles) of dormant (living) chains vs. particle diameter. The upper (chain) and lower (PDI) horizontal lines are the values in the corresponding bulk (not compartmentalized) system. Reprinted with permission from ref. 41. Copyright 2010 American Chemical Society. | ||
Thomson and Cunningham argue that the offset is caused by the fact that in ATRP, the activation rate increases with decreasing particle size as a consequence of the concentration of Cu(I) increasing (shown by simulations) as a result of the confined space effect on the deactivation reaction (in NMP, the activation rate is independent of particle size because it is a unimolecular reaction). However, the problem with this rationale is the following: a plot of log[P˙] (or log chain as in Fig. 10) vs. log d results in a straight line of slope 3 for sufficiently small particles. The reason for this behavior is that in this particle size regime, [P˙] is proportional to the time taken for deactivation to occur after an activation event (=NAvp/kdeact).25,37 As the particle size decreases, the deactivation time decreases with d3, and hence it can be shown that the slope = 3. Now, this is based on the activation rate being constant with decreasing particle size, i.e. the only factor that influences [P˙] is the deactivation time (rate of deactivation). If the increased rate of deactivation caused by the confined space effect with decreasing particle size was to cause a significant increase in activation rate via an increase in [Cu(I)], then the slope would not be equal to 3 (the slope would decrease with decreasing d). Thus, the explanation offered appears inconsistent with this aspect of the simulated data. It cannot, however, be excluded with absolute certainty that the increase in activation rate due to the confined space effect may be a factor in the region where the slope is not equal to 3 (in the region where an increase in Rp is seen), although this appears unlikely due to the relatively weak confined space effect in this region. One also wonders whether the fluctuation effect30 may play a role in causing the offset between Rp and Mw/Mn.
Whatever the rationale, the important fact remains that according to the simulations by Thomson and Cunningham,41 it is possible to achieve a simultaneous increase in Rp and the level of control (and livingness) in the case of ATRP. However, it remains to be clarified whether this is a special case, and to what extent the conclusions depend on whether control of the MWD is assessed viaN or viaMw/Mn of dormant chains.
Effects of diffusion-control
As the conversion increases, the viscosity within the polymer particles increases and at some point it is likely that the deactivation reaction will become diffusion-controlled, i.e.kdeact would thus decrease with increasing conversion beyond a certain conversion level. It is well-known that the termination reaction is diffusion-controlled from the outset of the polymerization, and kt thus depends on conversion.54,55 As such, it is expected that the effects of compartmentalization in both NMP and ATRP to some extent depend on the conversion level. All the theoretical studies described above have been concerned with low conversion polymerizations and such effects have therefore been deemed negligible.Zetterlund40 carried out simulations employing modified Smith–Ewart equations in connection with conversion-dependent rate coefficients kdeact and kt for the system styrene/polystyrene–Cl/CuCl/dNbpy2 at 75 °C. The decrease in kt with increasing conversion causes a minor increase in livingness at high conversion relative to when kt remains constant. The decrease in kdeact at high conversion results in an increase in the number of monomer units added per activation–deactivation cycle, and consequently slight loss of control (broader MWD) relative to when kdeact remains constant. The same qualitative effects are expected in the case of NMP.
Effect of exit of deactivator
In the discussions above, it has been assumed that the deactivator (nitroxide in NMP, Cu(II) complex in ATRP) is confined to a given particle/monomer droplet and is unable to undergo phase transition (exit to the aqueous phase). In a real system, the deactivator may partition between the organic and the aqueous phase, the extent of which is a function of its partitioning coefficient between the two phases. There are two distinct effects of this behavior on NMP/ATRP in a compartmentalized dispersed system: (i) partitioning of the deactivator means that the overall deactivator concentration in the organic phase is reduced; (ii) exit of a deactivator species from a particle (and possible subsequent entry into another particle) affects the polymerization in that particular particle, in particular if the confined space effect is operative in the system. The effect of partitioning in the absence of compartmentalization in NMP and ATRP is relatively well-understood and will not be described here.56–60Deactivator not compartmentalized
If the deactivator partitions between the two phases in such a way that it is able to move relatively freely between particles, only the propagating radicals are compartmentalized. In such a case, the segregation effect may be operative, but the deactivation reaction is not compartmentalized (no confined space effect). This case was analysed theoretically by Charleux for NMP,46 who derived eqn (11):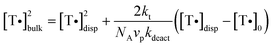 | (11) |
Confined space effect vs. deactivator exit
A complex situation arises in a system where the confined space effect is operative and the deactivator undergoes significant exit. The phase equilibrium times for styrene/TEMPO are significantly shorter than the time for deactivation to occur,57 suggesting that exit of nitroxide can compete with deactivation (this is also anticipated to apply to radicals generated in pairs by spontaneous initiation of St inside particles, and consequently some fraction of these radicals are likely to exit instead of undergoing geminate termination in small particles28,29).If we consider the simplest case (which is in fact common for systems where the confined space effect is operative) where an activation event occurs in a particle that contains no propagating radicals and no deactivator, the activation event generates a particle containing one propagating radical P˙ and one deactivator species (e.g. a nitroxide T˙). Next, one of two events may occur (we neglect propagation, which is not relevant here): T˙ may exit the particle to the continuous phase, or deactivation may occur. Under such conditions, assuming phase transfer equilibrium, the rate of exit of T˙ is given by eqn (12).1,47
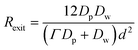 | (12) |
The rate of deactivation is dictated by:
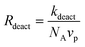 | (13) |
Fig. 11 shows the probability of exit as a function of diameter as given by Rexit/(Rexit + Rdeact) from eqn (12) and (13). The probability of exit of T˙ increases with increasing particle size, because the rate of deactivation is proportional to d−3 whereas the rate of exit is proportional to d−2. Even for the hydrophobic nitroxide TEMPO (partition coefficient = [TEMPO]org/[TEMPO]aq = Γ = 98.862), exit dominates over deactivation for particles with diameters over 20 nm. For the more water-soluble OH-TEMPO (Γ = 2.262), exit is the predominant event even for very small particles. Note, however, that this analysis is only valid for particles containing 1P˙ and 1T˙. As a result of the competition between deactivation and exit, the magnitude of the confined space effect (as manifested in reduction in Rp and lower Mw/Mn) decreases with increasing water-solubility of the deactivator. As will be shown in the next paragraph, exit of T˙ from a particle containing 1P˙ and 1T˙ can be detrimental with regard to control/livingness.
![Probabilities of nitroxide exit in a particle/monomer droplet containing one nitroxide and one propagating radical as functions of particle diameter for different nitroxide partition coefficients (Γ = [T˙]org/[T˙]aq) based on eqn (12) and (13). Reprinted with permission from ref. 34. Copyright 2010 Wiley-VCH Verlag GmbH & Co.](/image/article/2011/PY/c0py00247j/c0py00247j-f11.gif) | ||
| Fig. 11 Probabilities of nitroxide exit in a particle/monomer droplet containing one nitroxide and one propagating radical as functions of particle diameter for different nitroxide partition coefficients (Γ = [T˙]org/[T˙]aq) based on eqn (12) and (13). Reprinted with permission from ref. 34. Copyright 2010 Wiley-VCH Verlag GmbH & Co. | ||
Tobita and Yanase29 investigated the effect of exit using their Monte Carlo simulation approach. They considered an NMP system where in addition to alkoxyamine activation (which generates P˙ and T˙), single radicals are also generated in the system. A single radical generation event corresponds to either (i) an activation event occurring in a particle containing no P˙ and no T˙ followed by T˙ exit, or (ii) spontaneous radical generation from St, generating a pair of radicals, occurring in a particle containing no P˙ and no T˙, followed by exit of one of the two radicals generated. Simulated MWDs obtained for particle diameters of 50, 75 and 150 nm are displayed in Fig. 12, revealing how the MWD is monomodal for 75 and 150 nm, but bimodal for 50 nm. The low molecular weight peak of the bimodal MWD corresponds to polymer having formed by NMP. The rationale for the high molecular weight peak is that for the smallest particle size, some fraction of the single radical generation events would occur in particles containing no nitroxide, and consequently such radicals would grow rapidly to high molecular weight without deactivation. A single radical in a small particle would propagate until chain transfer to monomer occurs (although chain transfer to monomer was not part of the model), as normally happens in conventional (non-living) emulsion- and microemulsion polymerizations with sufficiently small particles where strong segregation of propagating radicals minimizes termination.63–65 The analysis presented in this section applies equally to NMP and ATRP.
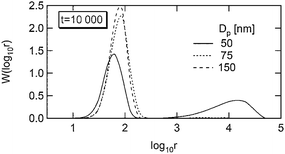 | ||
| Fig. 12 Molecular weight distributions obtained by Monte Carlo simulations for TEMPO-mediated radical polymerization (NMP) of styrene with single radical generation in the dispersed phase for different particle sizes as indicated (see text for details). Reprinted with permission from ref. 29. Copyright 2007 Wiley-VCH Verlag GmbH & Co. | ||
Experimental evidence of compartmentalization
Nitroxide-mediated radical polymerization
The most studied dispersed NMP system is by far TEMPO-mediated radical polymerization of St in miniemulsion, which in most cases behaves similarly to in bulk.66–72 The particle diameters in such systems generally tend to be in the range 80–200 nm, which according to simulations is too large for compartmentalization effects to be important.25,26 An additional factor is that particle size distributions in miniemulsions are often relatively broad,2,3,67 which would reduce any effect of compartmentalization as the predominant loci of polymerization (in terms of mass of polymer formed) would be the larger particles.There are many examples in the literature where Rp or other aspects of the polymerization in miniemulsion/microemulsion NMP are significantly different from the corresponding bulk system.27,28,33,73–86 However, dispersed systems may exhibit a range of features that are not present in bulk systems, and as such it is often difficult to ascribe specific differences in polymerization behavior to compartmentalization effects. For example, factors other than compartmentalization and nitroxide partitioning that may affect NMP in dispersed systems include the interface effect on deactivation,77,80,82–84,86 exit of small radicals (from chain transfer to monomer and/or spontaneous initiation of styrene),28 enhanced (spontaneous) radical generation,63,74,78 the Laplace pressure inside the particles,87 as well as effects related to the monomer concentration at the polymerization locus.
Perhaps the most compelling experimental evidence of compartmentalization effects in NMP is the results published by Cunningham and coworkers28 for the TEMPO-mediated miniemulsion polymerization of St at 135 °C. Fig. 13 depicts conversion–time data obtained in that study, showing how a decrease in the weight-average particle diameter from 180 to 50 nm (achieved by increasing the concentration of the surfactant Dowfax 8390) led to a marked reduction in Rp. The livingness of the system increased with decreasing particle size, but the level of control (Mw/Mn) was relatively unaffected by the particle size. The polymerization in bulk was faster than all miniemulsion polymerizations, but the bulk system exhibited intermediate livingness. The above experimental findings are largely consistent with compartmentalization effects. The reduction in Rp with decreasing particle size is consistent with the confined space effect on deactivation and geminate termination of radicals generated by spontaneous initiation of St, and the improved livingness is consistent with reduced levels of termination due to segregation.
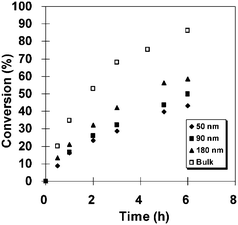 | ||
| Fig. 13 Conversion vs. time for TEMPO-mediated radical polymerization of styrene in bulk and miniemulsion for different particle sizes (different concentrations of the surfactant Dowfax 8390) at 135 °C. Reprinted with permission from ref. 28. Copyright 2009 American Chemical Society. | ||
Simulations corresponding to the specific conditions of the experimental data of Cunningham and coworkers28 in Fig. 13 were carried out in the present study (Fig. 14) based on the model employing the modified Smith–Ewart equations.25 Rate coefficients used: kp = 2950 M−1 s−1;88kt = 1.82 × 108 M−1 s−1;89kact = 0.004 s−1;90kdeact = 7.6 × 107 M−1 s−1;90,91ki,th = 3.91 × 10−10;50 initial polystyrene–TEMPO macroinitiator concentration = 0.0191 M. The simulated conversion–time data for the bulk system somewhat underestimated the experimentally obtained polymerization rate (note that the model for the compartmentalized system is based on the model for the bulk system, i.e. the outputs of the two models converge for sufficiently large particles). In order to obtain good agreement between the experimentally obtained miniemulsion data of particle diameter 50 nm, the simulations required a diameter of approximately 38 nm. Considering the numerous sources of error both in the experimental data (error in measurements of conversion and particle size) and simulations (ideal system assumed, i.e. all particles of same size, error in values of rate coefficients used, chain-length dependence of termination not accounted for), the agreement between model and experiment is in fact remarkably good, thus providing validation of the model (Fig. 14).
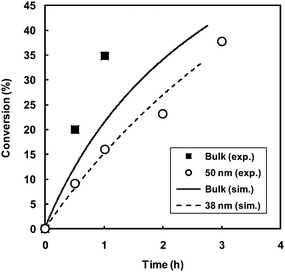 | ||
| Fig. 14 Conversion vs. time for TEMPO-mediated radical polymerization of styrene in bulk and miniemulsion for different particle diameters at 135 °C obtained by experiment (taken from ref. 28 and Fig. 13) and by simulations carried out in this work based on the model in ref. 25 with rate coefficients given in the text. | ||
The TEMPO/St system has also been studied in microemulsion. A microemulsion system5,6 is thermodynamically stable (unlike a miniemulsion,2 which requires energy input for its formation) and microemulsion polymerizations generally result in particle diameters in the range 10–60 nm, smaller than miniemulsion polymerizations, and are as such prime candidates for manifestation of compartmentalization effects. Okubo and Zetterlund27 reported that Rp was lower in microemulsion than bulk for NMP of styrene at 125 °C for both TEMPO (d = 44–68 nm) and SG1 (d = 21–27 nm), consistent with the confined space effect. Less pronounced retardation in the case of SG1 can be rationalized based on this nitroxide exhibiting higher water solubility than TEMPO, i.e. nitroxide exit (partitioning) would counteract the confined space effect on deactivation.
Microemulsion NMP of St at 100 °C has also been performed employing two nitroxides of different water solubilities (SG1: relatively high water solubility; TIPNO: relatively low water solubility) to investigate the influence of nitroxide exit.33Rp was lower in microemulsion than in bulk for both nitroxides, and the MWDs were significantly narrower in microemulsion than bulk at low conversion (Fig. 15), consistent with compartmentalization effects. However, the AIBN initiator efficiency was markedly lower in microemulsion than bulk, presumably due to the confined space effect on the geminate termination reaction of the initiator radicals. The lower initiator efficiency in microemulsion would result in a greater excess of nitroxide than in bulk, and this would also contribute towards lower Rp and narrower MWD in microemulsion. The extent of retardation relative to bulk was much less significant for the more water soluble nitroxide SG1 than for TIPNO, consistent with SG1 undergoing more exit.
![Molecular weight distributions (normalized to peak height) for TIPNO-mediated polymerization of styrene at 100 °C with [TIPNO]0/[AIBN]0 = 1.8 in microemulsion (full line) and bulk (dotted line). Microemulsion: Mn = 19 300 g mol−1, Mw/Mn = 1.30, 18% conv. (36 h). Bulk: Mn = 20 200 g mol−1, Mw/Mn = 1.56, 28% conv. (2 h). Reprinted with permission from ref. 33. Copyright 2009 American Chemical Society.](/image/article/2011/PY/c0py00247j/c0py00247j-f15.gif) | ||
Fig. 15 Molecular weight distributions (normalized to peak height) for TIPNO-mediated polymerization of styrene at 100 °C with [TIPNO]0/[AIBN]0 = 1.8 in microemulsion (full line) and bulk (dotted line). Microemulsion: Mn = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1, Mw/Mn = 1.30, 18% conv. (36 h). Bulk: Mn = 20200 g mol−1, Mw/Mn = 1.56, 28% conv. (2 h). Reprinted with permission from ref. 33. Copyright 2009 American Chemical Society. 300 g mol−1, Mw/Mn = 1.30, 18% conv. (36 h). Bulk: Mn = 20200 g mol−1, Mw/Mn = 1.56, 28% conv. (2 h). Reprinted with permission from ref. 33. Copyright 2009 American Chemical Society. | ||
Delaittre and Charleux investigated compartmentalization effects in the surfactant free SG1-mediated emulsion polymerization of St with particle formation occurring via the self-assembly mechanism relying on a poly(sodium acrylate) macroalkoxyamine acting as initiator and surfactant.47 It was concluded that the kinetics were not significantly affected by particle size, even though the particles were as small as 31–66 nm. It is, however, difficult to compare an emulsion polymerization in Interval II (monomer droplets present) with a bulk system because the monomer concentration in the polymer particles is not the same as in the corresponding bulk system at the same overall conversion. It was argued that there was no confined space effect on the deactivation due to the high water solubility of SG1. However, it is important to realize that there are two criteria that need to be fulfilled for the confined space effect to be operative: (i) the water solubility of the deactivator (SG1) is sufficiently low and (ii) the number of deactivator species per particle is sufficiently low as given by eqn (1). The average number of SG1 molecules per particle was reported to be in the range 45–300, and one can thus conclude immediately that there would be no confined space effect in this system, regardless of the ability of SG1 to diffuse between particles. As outlined earlier, smaller particles are required for compartmentalization effects to be important in NMP (and ATRP) systems with high equilibrium constants like SG1, and this would be one major reason that compartmentalization effects seem to manifest themselves more readily in TEMPO-based systems than SG1-based systems. To date, no convincing evidence of compartmentalization effects in SG1-mediated polymerizations in emulsion47,92 and miniemulsion73 have been reported. Slightly elevated Rp in miniemulsion compared to bulk has been observed in SG1-mediated polymerizations of styrene, attributed to segregation of propagating radicals and/or SG1 partitioning and/or decomposition of SG1 in the aqueous phase.75
Atom transfer radical polymerization
Although there now exists a large body of work on ATRP in dispersed systems,13,14 it is difficult to find data that provide clear evidence of compartmentalization effects. There are few reports where particle size effects are studied or where comparison is made with the corresponding bulk/solution polymerization. Matyjaszewski and coworkers93 found that for the reverse ATRP system n-butyl methacrylate/AIBN/CuBr2/dNbpy1/90 °C, there was no significant difference in polymerization behavior with (d = 235 nm) and without (d = 1070 nm) hexadecane despite very different particle sizes. Rp was higher in bulk than miniemulsion, but it is unlikely that the confined space effect on deactivation would be at play for such large particles, and it is considered more likely that the main factor was the higher initiation efficiency in bulk. The corresponding direct ATRP miniemulsion polymerization was also not significantly influenced by decreasing the particle size from d = 1.5 µm to 300 nm. Matyjaszewski and coworkers94 also studied the n-butyl methacrylate/CuBr2/BPMODA (so-called SR&NI system95) miniemulsion polymerization (d = 252 nm), and found no difference in polymerization behavior compared to bulk. Similar results in miniemulsion and bulk were also reported with n-butyl acrylate (d = 270 and 305 nm). The lack of apparent compartmentalization effects in all of the above investigations is broadly speaking consistent with theory in that smaller particles are needed for compartmentalization effects to play a significant role.37,39,41 In this regard, microemulsion ATRP is ideally suited for compartmentalization studies. Although microemulsion ATRP has been performed,96–98 the corresponding bulk systems were not reported, and it is therefore difficult to draw conclusions about compartmentalization effects.The only body of work that provides convincing experimental evidence of compartmentalization effects in ATRP is that of Simms and Cunningham.38,99,100 They carried out miniemulsion ATRP of n-butyl methacrylate with CuBr2/EHA6TREN and the redox initiating system hydrogen peroxide/ascorbic acid at 90 °C.38 This system essentially represents a hybrid between reverse ATRP and Activators Generated by Electron Transfer (AGET) ATRP.12,101 The particle diameters were varied between 119 and 212 nm by changing the amount of the cationic surfactant cetyltrimethylammonium bromide (CTAB, it was confirmed that the particle size, as opposed to the surfactant concentration, caused alterations in polymerization behavior). The polymerization rate decreased and the control over the MWD, as judged by Mw/Mn, improved with decreasing particle size (Fig. 16), consistent with the confined space effect on the deactivation reaction. Another interesting aspect is that a molecular weight as high as Mn = 989900 g mol−1 could be achieved, yet with good control (Mw/Mn = 1.25). Such high molecular weight cannot be reached with satisfactory control/livingness in the corresponding bulk/solution system (note that the corresponding bulk system cannot be investigated in this case due to the nature of the initiating system) due to the propagating radicals eventually undergoing termination, transfer or other side reactions if the cumulative active time of a single chain is as long as required for that degree of polymerization.17 This is also consistent with segregation suppressing termination, and the confined space effect resulting in control being maintained to unusually high degrees of polymerization. The nature of the initiation system may also be an important factor, given the fact that ascorbic acid will not only act as a redox initiator component with hydrogen peroxide, but also function as reducing agent converting Cu(II) to Cu(I) (the key step in AGET ATRP95).
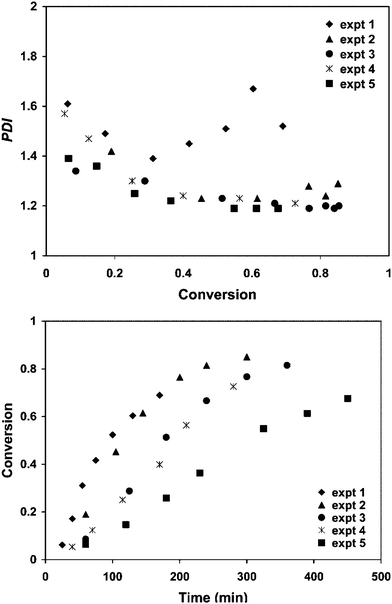 | ||
| Fig. 16 M w/Mn (PDI) and conversion vs. time data for miniemulsion ATRP of n-butyl methacrylate with CuBr2/EHA6TREN and the redox initiating system hydrogen peroxide/ascorbic acid at 90 °C for various particle diameters: expt 1: 212 nm; expt 2: 176 nm; expt 3: 142 nm; expt 4: 141 nm; and expt 5: 119 nm. Reprinted with permission from ref. 38. Copyright 2008 American Chemical Society. | ||
A frequent dilemma when trying to detect or exploit compartmentalization effects in miniemulsion is that it is normally necessary to use quite low concentrations of polymer chains (i.e. low alkoxyamine concentration in NMP, low alkylhalide concentration in ATRP), in other words high target molecular weights, to achieve sufficiently low values of propagating radicals and/or deactivator species per particle. This often in turn leads to impractically low polymerization rates. The ATRP system of Simms and Cunningham has very low concentrations of chains (thus high molecular weights), yet the polymerization rate is reasonably high. This has been speculated38 to be related to the initiation system hydrogen peroxide/ascorbic acid, which may result in an advantageous ratio of Cu(I)/Cu(II), a key factor probably being the presence of ascorbic acid leading to a low concentration of Cu(II) (thus high Rp).
Cobalt-mediated radical polmerization
The presence of certain low-spin Co(II) complexes such as {bis{µ-{(2,3-butanedione dioximato)(2)-O,O′}}tetrafluorodiborato(2-)-N,N′,N″,N‴}cobalt (COBF) in low concentration during polymerization of methacrylates results in generation of polymer with 2-carbalkoxy-2-propenyl end groups via catalytic chain transfer.102–104 Co(II) abstracts hydrogen from the α-methyl group of the propagating radical generating macromonomer and Co(III), and Co(III) subsequently undergoes hydrogen transfer with monomer to give Co(II) and a monomer radical, which reinitiates polymerization. In the case of styrene polymerization, Co(III)–C bonds are competitively generated. Co(III)–C bonds are weak, and the cleavage of Co(III)–C occurs readily, generating Co(II) and a carbon-center radical. In the case of acrylates, reversible formation of Co(III)–C bonds is the main reaction, resulting in the formation of a CLRP equilibrium based on the PRE effect.105–110 Thus, if such a polymerization was performed in a dispersed system, it is possible that compartmentalization effects may operate in terms of both the segregation effect on termination and the confined space effect on deactivation (i.e. formation of Co(III)–C bonds). Modeling and simulations of compartmentalization effects in cobalt-mediated radical polymerization (CMRP) have to date not been reported.Jerome and coworkers carried out CMRP of vinyl acetate (VAc) in aqueous suspension.111 The particle size (close to 1 mm diameter) was much too large for any compartmentalization effects to occur. Jerome and coworkers110 also reported CMRP of VAc in miniemulsion at low temperature (0–30 °C) using poly(VAc) macroinitiators end-capped by a cobalt acetylacetonate complex and 2,2′-azobis(4-methoxy-2,4-dimethyl valeronitrile) (V-70) generating particles with diameters near 100 nm. The rate of polymerization was exceptionally high, much higher than in bulk/solution CLRP of VAc, but good control/livingness was nonetheless obtained. Quantification of the partitioning of the deactivator Co(II)(acac)2 between the organic and aqueous phases revealed that excessive partitioning to the aqueous phase was occurring,111 which would counteract any confined space effect on deactivation. One can speculate that the segregation effect on termination combined with partitioning of the deactivator to the aqueous phase caused the high polymerization rate.
Final remarks and outlook
As outlined in this review, compartmentalization in nanoreactors of NMP and ATRP (or indeed any CLRP system that is based on the persistent radical effect, so long as the deactivator concentration is sufficiently low) offers a means of improving both the level of control over the MWD (narrower) and the degree of livingness (end-functionality), albeit usually, but not always, at the price of a reduction in polymerization rate. It is important to point out that such simultaneous improvements in both control and livingness cannot be achieved by simply diluting the corresponding bulk/solution system or by increasing the initial concentration of deactivator,36 further highlighting the future potential of the concept of compartmentalization in nanoreactors in polymer synthesis.One of the shortcomings of both NMP and ATRP, and indeed of all CLRP systems developed to date (with the possible exception of single-electron transfer living radical polymerization (SET-LRP)112), is that it is difficult to reach high molecular weight (>100000 g mol−1) while maintaining control and livingness. This is due to the fact that one of the premises of CLRP is that the cumulative time a chain spends in its active state (i.e. as a propagating radical as opposed to being dormant) is shorter than in a conventional non-living radical polymerization (assuming equal propagating radical concentrations in both systems), thus limiting the accessible molecular weights.17 Polymerization in nanoreactors offers an exciting solution to this problem, in that the segregation effect can significantly prolong the time a polymer chain can be “allowed” to be in its active state without the probability of termination or other side reactions becoming prohibitively high. This concept has recently been demonstrated experimentally in the case of ATRP as outlined above.
Another exciting future prospect of CLRP in nanoreactors is to exploit compartmentalization effects to improve systems that perform poorly or completely unsatisfactorily under homogeneous conditions in bulk/solution. An example might be the TEMPO-mediated radical polymerization of acrylates. Experimental work has shown that TEMPO-mediated acrylate polymerization does not proceed satisfactorily, postulated mainly to be due to excessive accumulation of free TEMPO. Recent theoretical work35 has indicated that it may be possible to at least partially overcome this problem by exploitation of compartmentalization effects in dispersed systems. Exploitation of compartmentalization effects essentially enables one to reduce the effective termination rate coefficient and increase the effective deactivation rate coefficient, thus providing a novel avenue (as opposed to chemical means, e.g. developing new nitroxides in NMP or ligands in ATRP) to develop and improve new systems.
Acknowledgements
PBZ is grateful to the Australian Research Council for financial support in the form of a Discovery Grant (DP1093343).References
- R. G. Gilbert, Emulsion Polymerization: A Mechanistic Approach, Academic Press, London, 1995 Search PubMed.
- J. M. Asua, Prog. Polym. Sci., 2002, 27, 1283 CrossRef CAS.
- K. Landfester, Macromol. Rapid Commun., 2001, 22, 896 CrossRef.
- F. J. Schork, Y. W. Luo, W. Smulders, J. P. Russum, A. Butte and K. Fontenot, Adv. Polym. Sci., 2005, 175, 129–255 CAS.
- F. Candau, Microemulsion Polymerization, In Polymeric Dispersions: Principles and Applications, ed. J. M. Asua, Kluwer Academic Publishers, 1997, pp. 127–140 Search PubMed.
- P. Y. Chow and L. M. Gan, Adv. Polym. Sci., 2005, 175, 257–298 CAS.
- K. E. J. Barrett, Dispersion Polymerization in Organic Media, Wiley, London, 1975 Search PubMed.
- J. L. Cawse, Dispersion Polymerization, in Emulsion Polymerization and Emulsion Polymers, ed. P. A. Lovell and M. S. El-Aasser, John Wiley & Sons Ltd., West Sussex, 1997, pp. 743–761 Search PubMed.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543–563 CrossRef CAS.
- P. B. Zetterlund, F. Aldabbagh and M. Okubo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3711–3728 CrossRef CAS.
- M. J. Monteiro, Macromolecules, 2010, 43, 1159–1168 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS.
- M. F. Cunningham, Prog. Polym. Sci., 2008, 33, 365–398 CrossRef CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS.
- W. Tang, T. Fukuda and K. Matyjaszewski, Macromolecules, 2006, 39, 4332–4337 CrossRef CAS.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, 2008 Search PubMed.
- H. Fischer, Criteria for Livingness and Control in Nitroxide-Mediated and Related Radical Polymerizations, in Advances in Controlled/Living Radical Polymerization, ed. K. Matyjaszewski, ACS Symposium Series 854, Washington, DC, 2003 Search PubMed.
- P. B. Zetterlund and M. Okubo, Macromolecules, 2006, 39, 8959–8967 CrossRef CAS.
- P. B. Zetterlund and M. Okubo, Macromol. Theory Simul., 2007, 16, 221–226 CrossRef CAS.
- J. Wakamatsu, M. Kawasaki, P. B. Zetterlund and M. Okubo, Macromol. Rapid Commun., 2007, 28, 2346–2353 CrossRef CAS.
- H. Maehata, C. Buragina, M. Cunningham and B. Keoshkerian, Macromolecules, 2007, 40, 7126–7131 CrossRef CAS.
- H. Tobita and F. Yanase, Macromol. Theory Simul., 2007, 16, 476–488 CrossRef CAS.
- H. Tobita, Macromol. Theory Simul., 2007, 16, 810–823 CrossRef CAS.
- H. Tobita, Macromol. Symp., 2008, 261, 36–45 CrossRef CAS.
- P. B. Zetterlund and M. Okubo, Macromol. Theory Simul., 2009, 18, 277–286 CrossRef CAS.
- P. B. Zetterlund, J. Wakamatsu and M. Okubo, Macromolecules, 2009, 42, 6944–6952 CrossRef CAS.
- P. B. Zetterlund, Macromol. Theory Simul., 2010, 19, 11–23 CAS.
- P. B. Zetterlund, Macromol. React. Eng. DOI:10.1002/mren.201000022.
- P. B. Zetterlund, Aust. J. Chem., 2010, 63, 1195–1200 CrossRef CAS.
- Y. Kagawa, P. B. Zetterlund, H. Minami and M. Okubo, Macromol. Theory Simul., 2006, 15, 608–613 CrossRef CAS.
- R. W. Simms and M. F. Cunningham, Macromolecules, 2008, 41, 5148–5155 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Macromolecules, 2009, 42, 2488–2496 CrossRef CAS.
- P. B. Zetterlund, Macromolecules, 2010, 43, 1387–1395 CrossRef CAS.
- M. E. Thomson and M. F. Cunningham, Macromolecules, 2010, 43, 2772–2779 CrossRef CAS.
- Y. Luo, R. Wang, L. Yang, B. Li and S. Zhu, Macromolecules, 2006, 39, 1328–1337 CrossRef CAS.
- H. Tobita, Macromol. Theory Simul., 2009, 18, 108–119 CrossRef CAS.
- H. Tobita, Macromol. Theory Simul., 2009, 18, 120–126 CrossRef CAS.
- H. Tobita, Macromol. Theory Simul., 2010, 288, 16–24 CAS.
- B. Charleux, Macromolecules, 2000, 33, 5358–5365 CrossRef CAS.
- G. Delaittre and B. Charleux, Macromolecules, 2008, 41, 2361–2367 CrossRef CAS.
- W. V. Smith and R. H. Ewart, J. Chem. Phys., 1948, 16, 592–599 CrossRef.
- A. Butte, G. Storti and M. Morbidelli, DECHEMA Monogr., 1998, 134, 497–507 Search PubMed.
- A. W. Hui and A. E. Hamielec, J. Appl. Polym. Sci., 1972, 16, 749–769 CrossRef CAS.
- K. S. Khuong, W. H. Jones and W. A. Pryor, J. Am. Chem. Soc., 2005, 127, 1265–1277 CrossRef CAS.
- H. Tobita, Y. Takada and M. Nomura, Macromolecules, 1994, 27, 3804–3811 CrossRef CAS.
- P. Taylor, Adv. Colloid Interface Sci., 1998, 75, 107–163 CrossRef CAS.
- M. Buback, M. Egorov, R. G. Gilbert, V. Kaminsky, O. F. Olaj, G. T. Russell, P. Vana and G. Zifferer, Macromol. Chem. Phys., 2002, 203, 2570–2582 CrossRef CAS.
- P. B. Zetterlund, H. Yamazoe, B. Yamada, D. J. T. Hill and P. J. Pomery, Macromolecules, 2001, 34, 7686–7691 CrossRef CAS.
- J. W. Ma, M. F. Cunningham, K. B. McAuley, B. Keoshkerian and M. K. Georges, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1081–1089 CrossRef CAS.
- J. W. Ma, M. F. Cunningham, K. B. McAuley, B. Keoshkerian and M. K. Georges, Macromol. Theory Simul., 2002, 11, 953–960 CrossRef CAS.
- J. W. Ma, M. F. Cunningham, K. B. McAuley, B. Keoshkerian and M. K. Georges, Chem. Eng. Sci., 2003, 58, 1163–1176 CrossRef CAS.
- P. B. Zetterlund and M. Okubo, Macromol. Theory Simul., 2005, 14, 415–420 CrossRef CAS.
- Y. Kagawa, P. B. Zetterlund, H. Minami and M. Okubo, Macromolecules, 2007, 40, 3062–3069 CrossRef CAS.
- K. Y. van Berkel, G. T. Russell and R. G. Gilbert, Macromolecules, 2003, 36, 3921–3931 CrossRef CAS.
- J. W. Ma, M. F. Cunningham, K. B. McAuley, B. Keoshkerian and M. K. Georges, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1081 CrossRef CAS.
- M. N. Alam, P. B. Zetterlund and M. Okubo, Polymer, 2008, 49, 883–892 CrossRef CAS.
- C. C. Co, P. Cotts, S. Burauer, R. de Vries and E. W. Kaler, Macromolecules, 2001, 34, 3245–3254 CrossRef CAS.
- M. Nomura and K. Suzuki, Macromol. Chem. Phys., 1997, 198, 3025–3039 CrossRef CAS.
- T. Prodpran, V. L. Dimonie, E. D. Sudol and M. S. El-Aasser, Macromol. Symp., 2000, 155, 1–14 CrossRef CAS.
- G. Pan, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Macromolecules, 2001, 34, 481–488 CrossRef CAS.
- B. Keoshkerian, P. J. MacLeod and M. K. Georges, Macromolecules, 2001, 34, 3594–3599 CrossRef CAS.
- G. Pan, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Macromolecules, 2002, 35, 6915–6919 CrossRef CAS.
- M. F. Cunningham, K. Tortosa, J. W. Ma and K. B. McAuley, Macromol. Symp., 2002, 182, 273–282 CrossRef CAS.
- M. F. Cunningham, M. Xie, K. B. McAuley, B. Keoshkerian and M. K. Georges, Macromolecules, 2002, 35, 59–66 CrossRef CAS.
- M. Cunningham, M. Lin, C. Buragina, S. Milton, D. Ng, C. C. Hsu and B. Keoshkerian, Polymer, 2005, 46, 1025–1032 CrossRef CAS.
- M. Lansalot, C. Farcet, B. Charleux, J. P. Vairon, R. Pirri and P. Tordo, Nitroxide-Mediated Controlled Free-Radical Emulsion and Miniemulsion Polymerizations of Styrene, in Controlled/Living Radical Polymerization: Progress in ATRP, NMP and RAFT, ed. K. Matyjaszewski, American Chemical Society, 2000, pp. 138–151 Search PubMed.
- M. F. Cunningham, M. Lin and B. Keoshkerian, JCT Res., 2004, 1, 33–39 Search PubMed.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2004, 37, 4453–4463 CrossRef CAS.
- P. B. Zetterlund, Md. N. Alam, H. Minami and M. Okubo, Macromol. Rapid Commun., 2005, 26, 955–960 CrossRef CAS.
- T. Nakamura, P. B. Zetterlund and M. Okubo, Macromol. Rapid Commun., 2006, 27, 2014–2018 CrossRef CAS.
- M. Lin, J. C. C. Hsu and M. F. Cunningham, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5974–5986 CrossRef CAS.
- Md. N. Alam, P. B. Zetterlund and M. Okubo, Macromol. Chem. Phys., 2006, 207, 1732–1741 CrossRef CAS.
- Md. N. Alam, P. B. Zetterlund and M. Okubo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4995–5004 CrossRef CAS.
- Y. Saka, P. B. Zetterlund and M. Okubo, Polymer, 2007, 48, 1229–1236 CrossRef CAS.
- P. B. Zetterlund, T. Nakamura and M. Okubo, Macromolecules, 2007, 40, 8663–8672 CrossRef CAS.
- Md. N. Alam, P. B. Zetterlund and M. Okubo, Polymer, 2008, 49, 3428–3435 CrossRef CAS.
- Md. N. Alam, P. B. Zetterlund and M. Okubo, Polymer, 2009, 50, 1632–1636 CrossRef CAS.
- P. B. Zetterlund, Y. Saka and M. Okubo, Macromol. Chem. Phys., 2009, 210, 140–149 CAS.
- P. B. Zetterlund, N. Alam and M. Okubo, Polymer, 2009, 50, 5661–5667 CrossRef CAS.
- P. B. Zetterlund and M. Okubo, Macromol. Theory Simul., 2006, 15, 40–45 CrossRef CAS.
- M. Buback, R. G. Gilbert, R. A. Hutchinson, B. Klumperman, F. D. Kuchta, B. G. Manders, K. F. O'Driscoll, G. T. Russell and J. Schweer, Macromol. Chem. Phys., 1995, 196, 3267–3280 CrossRef CAS.
- M. Buback, C. Kowollik, C. Kurz and A. Wahl, Macromol. Chem. Phys., 2000, 201, 464–469 CrossRef CAS.
- A. Goto, T. Terauchi, T. Fukuda and T. Miyamoto, Macromol. Rapid Commun., 1997, 18, 673–681 CAS.
- T. Fukuda, T. Terauchi, A. Goto, K. Ohno, Y. Tsujii, T. Miyamoto, S. Kobatake and B. Yamada, Macromolecules, 1996, 29, 6393–6398 CrossRef CAS.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2005, 38, 9963–9973 CrossRef CAS.
- K. Matyjaszewski, J. Qiu, N. V. Tsarevsky and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4724–4734 CrossRef CAS.
- M. Li and K. Matyjaszewski, Macromolecules, 2004, 37, 2106–2112 CrossRef CAS.
- J. Gromada and K. Matyjaszewski, Macromolecules, 2001, 34, 7664–7671 CrossRef CAS.
- K. Min and K. Matyjaszewski, Macromolecules, 2005, 38, 8131–8134 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 10521–10526 CrossRef CAS.
- Y. Kagawa, M. Kawasaki, P. B. Zetterlund and M. Okubo, Macromol. Rapid Commun., 2007, 28, 2354–2360 CrossRef CAS.
- R. W. Simms and M. F. Cunningham, Macromolecules, 2007, 40, 860–866 CrossRef CAS.
- R. W. Simms and M. F. Cunningham, Macromol. Symp., 2008, 261, 32–35 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2005, 127, 3825–3830 CrossRef CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611–3659 CrossRef CAS.
- J. P. A. Heuts, G. E. Roberts and J. D. Biasutti, Aust. J. Chem., 2002, 55, 381–398 CrossRef CAS.
- B. Yamada, P. B. Zetterlund and E. Sato, Prog. Polym. Sci., 2006, 31, 835–877 CrossRef CAS.
- B. B. Wayland, G. Poszmik, S. Mukerjee and M. Fryd, J. Am. Chem. Soc., 1994, 116, 7943–7944 CrossRef CAS.
- B. B. Wayland, L. Basickes, S. Mukerjee, M. Wei and M. Fryd, Macromolecules, 1997, 30, 8109–8112 CrossRef CAS.
- Z. Lu, M. Fryd and B. B. Wayland, Macromolecules, 2004, 37, 2686–2687 CrossRef CAS.
- A. Debuigne, J. R. Caille and R. Jerome, Angew. Chem., Int. Ed., 2005, 44, 1101–1104 CrossRef CAS.
- B. B. Wayland, C. H. Peng, X. Fu, Z. Lu and M. Fryd, Macromolecules, 2006, 39, 8219–8222 CrossRef CAS.
- C. Detrembleur, A. Debuigne, R. Bryaskova, B. Charleux and R. Jerome, Macromol. Rapid Commun., 2006, 27, 37–41 CrossRef CAS.
- A. Debuigne, J. R. Caille, C. Detrembleur and R. Jerome, Angew. Chem., Int. Ed., 2005, 44, 3439–3442 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |