Oligoagarose-g-polycaprolactone loaded nanoparticles for drug delivery applications†
Archana
Bhaw-Luximon
a,
Laila Musharat
Meeram
a,
Yeshma
Jugdawa
a,
William
Helbert
b and
Dhanjay
Jhurry
*a
aDepartment of Chemistry, Faculty of Science, Réduit, Mauritius. E-mail: djhurry@uom.ac.mu; Fax: +230-4656825; Tel: +230-4037439
bEquipe Structure des Polysaccharides Marins, UMR 7139 (CNRS-UPMC) Végétaux marins et Biomolécules, Station Biologique, 29682, Roscoff, France
First published on 29th October 2010
Abstract
Well-defined oligoagarose-g-polycaprolactone copolymers of varying PCL chain lengths have been synthesised using protection/deprotection chemistry. The graft copolymers showed amphiphilic behaviour with spherical micelles in the size range 10 to 20 nm. Ketoprofen was loaded into the core of the micelles. The drug loading efficiency was shown to increase with the length of the hydrophobic PCL chain: 2% for PCL10 and 5.5% for PCL15. Sustained drug release was observed over a period of 72 h and was faster with shorter PCL chains.
The design of nano-drug delivery devices requires the consideration of a number of important factors amongst which are toxicity and biodegradability. Polysaccharides offer the critical advantages of being highly stable, non-toxic, hydrophilic and biodegradable. The use of native polysaccharides as controlled delivery systems has attracted a lot of interest. Due to their biodegradation and solubility in aqueous medium, they are suitable for bioerodible drug delivery systems. Indeed the rate of drug release was found to increase with the use of a water-soluble polymer as a major constituent.1 In addition, the presence of hydroxyl and/or amino groups present on polysaccharides can be further used for chemical modification.2 The hydroxyl groups also allow for specific mucoadhesion or receptor recognition. Natural polysaccharides have been combined with synthetic polymers to yield novel block and graft copolymers for biomedical applications. Only a few such block polysaccharide copolymers have been reported to form micelles in water and they have all been synthesised from dextrans.3–7 Polysaccharide-g-polyesters forming micelles have been reported using mainly dextran,8 chitosan9–16 and cellulose.17 However, agarose-g-polyesters as drug delivery systems have not been reported yet. Agarose, a natural polysaccharide, is extracted from red algae species and is commonly used in the biomedical field. Agarose beads,18agarose hydrogel nanoparticles,19glutaraldehyde cross-linked agarose20 and agarose-g-hyaluronan microparticles21 have been used for drug encapsulation.
Agarose-based systems present the unique advantage of having galactose end groups which can play a key role in cell targeting.22–25 We have recently reported on the grafting of polycaprolactone onto oligoagarose, obtained via controlled enzymatic degradation of native agarose.26,27 We report here on the solution properties of these copolymers and provide an assessment of their efficiency as nano drug carrier system using anti-inflammatory ketoprofen as model drug.
DSC and SEM characterisation of oligoagarose-g-PCL
Partially acetylated oligoagarose was used as macroinitiator in the presence of Sn(Oct)2 as catalyst for the ring-opening polymerisation of ε-caprolactone.26PCL of varying chain lengths were thus successfully grafted onto oligoagarose, as confirmed by NMR and SEC. DSC analysis of oligoagarose shows a broad endotherm centred at 85 °C while the graft copolymer with free hydroxyl groups exhibit a relatively sharper transition with three peaks in the range of 41.5–51.3 °C (ESI†). DSC analysis of a physical mixture of oligoagarose and PCL10 gave a higher melting transition at 53.8–56.3 °C (ESI†). The melting transition of the graft copolymer can therefore only be attributed to the PCL segments. The Tm value recorded is found to be lower than that of PCL homopolymer (60 °C). Similar results have been reported for chitosan-g-polycaprolactone copolymer with the presence of two peaks in the DSC profile at 51.8 and 56.7 °C.28 Another study performed on chitosan-g-polycaprolactone showed a fall in Tm to 45 °C depending on the ratio of polycaprolactone chain grafted.11 The broadness of the peak can be attributed to the difference in number of grafts per oligoagarose chain.Scanning electron microscopy (SEM) micrographs of starting oligoagarose, acetylated oligoagarose, and oligoagarose-g-PCL are shown in Fig. 1. Pure oligoagarose shows a layered structure similarly to reported SEM of high molar mass chitosan16 but upon derivatisation with acetic anhydride, spherical granules are observed due to loss in crystallinity (Fig. 1b). After the grafting process, another change of morphology can be noted (Fig. 1c), as a sign of successful grafting. This observation was also reported previously in the case of chitin-g-PCL.29 To further confirm the grafting, the SEM micrograph of a physical mixture of oligoAga and PCL10 was recorded (Fig. 1d) and reflects more of a heterogeneous mixture as compared to Fig. 1c.
 | ||
| Fig. 1 SEM micrographs of (a) oligoAga (b) AcO-oligoAga (c) oligoAga-g-PCL (d) physical mixture of oligoAga and PCL. | ||
Solution properties and drug loading/release
In water, the copolymers self-assemble into micelles with narrow particle size distributions (Fig. 2) and with an average size of 10 to 20 nm as determined by DLS. CMC was measured by DLS (ESI†) and found to be in the range of 2.7E-5 mmol mL−1. TEM images showed the presence of star-shaped spherical micelles (Fig. 3) with mean diameter 12 nm for HO-OligoAga-g-PCL10 and 8 nm for HO-OligoAga-g-PCL15. The smaller average size as compared to DLS measurement is probably due to the dry state of the TEM measurement.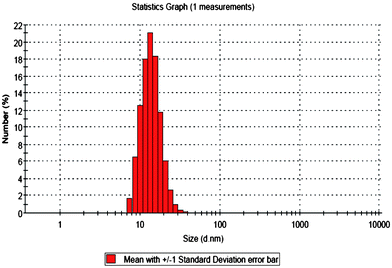 | ||
| Fig. 2 Particle size distribution of HO-OligoAgo-g-PCL10 using DLS measurement (concentration = 0.5 mg ml−1 in water). | ||
 | ||
| Fig. 3 TEM images of (i, ii) HO-OligoAga-g-PCL10 and (iii, iv) HO-OligoAga-g-PCL15. | ||
Ketoprofen was used as a model drug for encapsulation studies. It has been used in a number of nanocarrier systems such as acrylic polymers,30 polyions/gelatin,31 cellulose ether polymers,32PLGA,33β-cyclodextrin,34 and Eudragit®L100.35 Its incorporation can occur via ionic interaction, hydrophobic interaction, hydrogen bonding or physical entrapment.
Incorporation of ketoprofen into the graft copolymers was performed through the acetone volatilisation method. The amount of drug incorporated was determined by UV spectroscopy. Ketoprofen solution showed a strong absorbance peak at 260 nm (ε260 = 15 000 M−1 cm−1), a benzoin type transition (S0 → S2 transition of π,π* nature) which is due to its aromatic ring while a solution of HO-oligoAga-g-PCL showed no signal in that region.
Thus, using the latter as the baseline, the UV spectrum of ketoprofen-loaded copolymer can be recorded (ESI†). The maximum drug loading which can be achieved with this graft copolymer was determined for two PCL chain lengths using eqn (1). PCL15 gave a maximum loading at 8 mg of drug while for PCL10 the maximum loading was at 6 mg of ketoprofen (Fig. 4). The difference in hydrophobicity could account for differences in interaction strength between ketoprofen and the polylactone, as explained by Lin et al.36
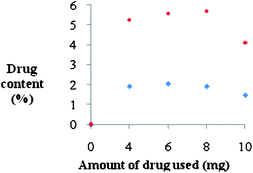 | ||
| Fig. 4 Drug loading profile of (♦) HO-OligoAga-g-PCL10 and (•) HO-OligoAgo-g-PCL15, both at a fixed copolymer concentration of 1 mg ml−1. | ||
The drug release profile was recorded in PBS at 37 °C. Sustained release was observed up to 60 h (Fig. 5).
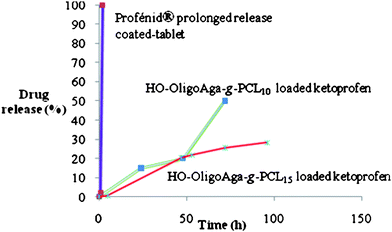
Interestingly, commercially available slow release Ketoprofen profenid® tablets with a cellulose coating showed release over just a few hours under identical conditions. The slowest release was observed from micelles with higher molar ratio of lactone monomer, here PCL15. This result confirms that release of drug is controlled by the hydrophobic polylactone chain length of micelles, as also found previously for chitosan-g-polydioxane copolymers16 exhibiting a diffusion-controlled release of drug.
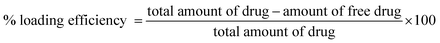 | (1) |
Conclusion
Oligoagarose-g-polycaprolactone copolymers of variable hydrophobic chain lengths have been synthesised and characterised. Hydrophobic Ketoprofen drug was successfully loaded into the hydrophobic core of the nanosized amphiphilic micelles. Both the loading capacity and drug release were shown to be dependent on the polycaprolactone chain length. A more sustained release was observed for the oligoagarose-g-PCL system compared to commercial Profenid tablets.References
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60(15), 1650 CrossRef CAS.
- P. Giunchedi, I. Genta, B. Conti, R. A. A. Muzzarelli and U. Conte, Biomaterials, 1998, 19(1), 157 CrossRef CAS.
- J.-Y. Liu and L.-M. Zhang, Carbohydr. Polym., 2007, 69, 196 CrossRef CAS.
- C. Houga, J.-F. Le Mains, R. Borsali, D. Taton and Y. Gnanou, Chem. Commun., 2007,(29), 3063 RSC.
- C. Houga, J. Giermanska, S. Lecommandoux, R. Borsali, D. Taton, Y. Gnanou and J.-F. Le Meins, Biomacromolecules, 2009, 10, 32 CrossRef CAS.
- O. S. Hernandez, G. M. Solima and F. M. Winnik, Polymer, 2007, 48, 921 CrossRef CAS.
- H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848 CrossRef CAS.
- R. Gref, J. Rodrigues and P. Couvreur, Macromolecules, 2002, 35(27), 9861 CrossRef CAS.
- Y. Liu, F. Tian and K. A. Hu, Carbohydr. Res., 2004, 339, 845 CrossRef CAS.
- Y. Wu, Y. Zheng, W. Yang, C. Wang, J. Hu and S. Fu, Carbohydr. Polym., 2005, 59(2), 165 CrossRef CAS.
- C. Wang, G. Li, S. Tao, R. Guo and Z. Yan, Carbohydr. Polym., 2006, 64(3), 466 CrossRef CAS.
- H. Feng and C.-M. Dong, Biomacromolecules, 2006, 7(11), 3069 CrossRef CAS.
- N. Bhattarai, H. R. Ramay, S.-H. Chou and M. Zhang, Int. J. Nanomed., 2006, 1(2), 181–187 CrossRef CAS.
- X. Guan, D. Quan, X. Shuai, K. Liao and K. Mai, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(12), 2556 CrossRef CAS.
- G. Li, Y. Zhuang, Q. Mu, M. Wang and Y. Fang, Carbohydr. Polym., 2008, 72, 60 CrossRef CAS.
- X. L. Wang, Y. Huang, J. Zhu, Y. B. Pan, R. He and Y. Z. Wang, Carbohydr. Res., 2009, 344(6), 801 CrossRef CAS.
- H. Dong, Q. Xu, Y. Li, S. Mo, S. Cai and L. Liu, Colloids Surf., B, 2008, 66(1), 26 CrossRef CAS.
- B. O. Häglund, S. M. Upadrashta, S. H. Neau and M. A. Cutrera, Drug Dev. Ind. Pharm., 1994, 20(6), 947 CrossRef CAS.
- N. Wang and X.-S. Wu, Pharm. Dev. Technol., 1997, 2(2), 135 CrossRef CAS.
- E. Yalçın and K. Çavuşoğlu, Turk. J. Biochem., 2008, 33(4), 148 Search PubMed.
- J.-Y. Huang, L. Bao, X. Mao and S.-Q. Tang, J. Appl. Polym. Sci., 2010, 117, 3568 CAS.
- J. Kopeček and R. Duncan, J. Controlled Release, 1987, 6(1), 315 CrossRef CAS.
- P. J. Julyan, L. W. Seymour, D. R. Ferry, S. Daryani, C. Boivin, J. Doranet, M. David, S. Hesslewood and D. J. Kerr, J. Controlled Release, 1999, 57, 281 CrossRef CAS.
- K. Yasugi, T. Nakamura, Y. Nagasaki, M. Kato and K. Kataoka, Macromolecules, 1999, 32, 8024 CrossRef CAS.
- Q. Wang, L. Zhang, H. Wei, Z.-H. Hu, Y. Y. Bei, J.-Y. Xu, W.-J. Wang, X.-N. Zhang and Q. Zhang, Nanomed.: Nanotechnol., Biol. Med., 2010, 6(2), 371 CrossRef CAS.
- A. Bhaw-Luximon, D. Jhurry, M. Heerah Booluck, W. Helbert and S. Genicot, Macromol. Symp., 2009, 277(1), 14 CrossRef CAS.
- M. Jam, D. Flament, J. Allouch, P. Potin, L. Thion, B. Kloareg, M. Czjzek, W. Helbert, G. Michel and T. Barbeyron, Biochem. J., 2005, 385, 703 CrossRef CAS.
- L. Liu, Y. Wang, X. Shen and Y. Fang, Biopolymers, 2005, 78, 163 CrossRef CAS.
- R. Jayakumar and H. Tamura, Int. J. Biol. Macromol., 2008, 43(1), 32 CrossRef CAS.
- H. Eerikäinen, L. Peltonen, J. Raula, J. Hirvonen and E. I. Kauppinen, AAPS PharmSciTech, 2004, 5(4), 19.
- A. I. Arida and M. M. Al-Tabakha, Eur. J. Pharm. Biopharm., 2007, 66, 48 CrossRef CAS.
- M. L. Vueba, L. A. E. Batista de Carvalho, F. Veiga, J. J. Sousa and M. E. Pina, Eur. J. Pharm. Biopharm., 2004, 58, 51 CrossRef CAS.
- P. Blasi, A. Schoubben, S. Giovagnoli, L. Perioli, M. Ricci and C. Rossi, AAPS PharmSciTech, 2007, 8(2), E1–E7 CrossRef.
- P. T. Tayade and P. R. Vavia, Indian J. Pharm. Sci., 2006, 68, 164 Search PubMed.
- R. Yang, Y. Wang, X. Zheng, J. Meng and X. Tang, Drug Dev. Ind. Pharm., 2008, 34, 83 CrossRef CAS.
- W.-J. Lin, C.-L. Wang and Y.-C. Chen, Drug Delivery, 2005, 12, 223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis and characterisation; statistic distribution calculated from TEM. See DOI: 10.1039/c0py00311e |
| This journal is © The Royal Society of Chemistry 2011 |