RAFT polymerization and thermal behavior of trimethylphosphonium polystyrenes for anion exchange membranes†
Kristoffer K.
Stokes‡
*,
Joshua A.
Orlicki
and
Frederick L.
Beyer
*
US Army Research Laboratory, Materials & Manufacturing Sciences Division, Aberdeen Proving Ground, Maryland, USA. E-mail: rick.beyer@us.army.mil; kstokes@celgard.com
First published on 15th November 2010
Abstract
Ionomers of trimethyl(4-vinylbenzyl)phosphonium chloride and styrene comonomers were synthesized with between 15 and 98 mol% ionic content. The thermal properties were explored and a significant increase in the glass transition of the high ion content polymers was found, most likely due to ion aggregation into clusters.
Alkaline fuel cells (AFCs) present an attractive approach to fuel cells for portable power. Although proton exchange membrane (PEM) fuel cells have substantial research initiatives embracing the technology, the nature of the electrochemical reaction in a PEM mandates the use of expensive platinum catalysts.1 By virtue of their alkaline environment, the cathodic reaction in an AFC proceeds much more rapidly and remains efficient while using less expensive nickel-based catalysts. Even though these catalyst materials can decrease cost and increase efficiency over those of PEM fuel cells, the electrolyte material required for AFCs has prevented the technology from achieving widespread commercial adoption.
Currently, the most useable AFC electrolyte materials are the same as those used in AFCs designed for the US space program, ca. 1965. Circulating electrolyte baths2 or asbestos matrices soaked in a concentrated potassium hydroxide solution are the electrolytes of choice. These arrangements suffer from two major drawbacks. First, aqueous electrolyte solutions add bulk and weight. Second, alkaline fuel cells can be poisoned when an impure fuel feed is introduced. Carbon dioxide impurities can chemically combine with the electrolyte medium, a reaction that can either simply decrease the concentration of active mobile electrolytes or create physical blockages through the precipitation of potassium carbonate. Both outcomes decrease the effective porosity of the matrix medium and thus the efficiency of the overall cell.3,4 A polymer electrolyte membrane that can transport hydroxide ions and is stable to alkaline conditions could accelerate the implementation of AFCs by mitigating these problems.
Ionomers are a class of polymeric materials that may be useful as anion exchange membranes. Incorporation of ion-containing monomers into a polymer backbone generally results in a material strongly driven to form a microphase segregated morphology of ionic aggregates in an uncharged matrix. Organization of the ionic aggregates into continuous channels may provide transport properties enabling the use of the ionomer as an anion exchange membrane. When appropriately functionalized, these channels would provide diffusion pathways for ion migration useful for fuel cell membranes, as well as other applications of semipermeable membrane technology.
Previous efforts toward anion exchange membranes include cationic ionomers containing quaternary ammonium5,6 and phosphonium7,8 salts as cation channels for anion transport. Here, we report the synthesis of a trimethylphosphonium chloride functionalized polystyrenevia reversible addition–fragmentation chain transfer (RAFT) polymerization, and the thermal behavior of these materials.
To deconvolute the effects of molecular weight and ion content, a controlled radical polymerization was used to synthesize the phosphonium-containing polystyrenes. Using living radical methodologies allows for control over molecular weight, molecular weight distribution, topology, and composition of polymers.9RAFT polymerization was chosen for the wide range of monomer classes that can be polymerized and its tolerance to many functional groups.9 Wang and Lowe utilized a similar pathway to synthesize a series of copolymer ampholytes containing phosphonium chloride and benzoic acid functionalities.10
Scheme 1 outlines the synthetic pathway to the cationic polystyrene. RAFT chain transfer agent (CTA) 1 was chosen for its diacid functionality. This highly polar RAFT CTA is soluble in a wide variety of solvents, both aqueous and organic, and useful for polymerizing styrenic systems with a controlled rate.10 Synthesis of trimethyl(4-vinylbenzyl)phosphonium chloride (2) is facile with a quantitative yield over three days stirring at room temperature. The material quickly precipitates and is readily filtered from the solvent to recover 2.
 | ||
| Scheme 1 Synthetic pathway to phosphonium functional polystyrene. | ||
Polymers 3a–f were synthesized under typical RAFT conditions using a diazo initiator. Methanol was found to be a suitable solvent for the reaction as it provides solubility of both 2 and the produced polymer,11,12 particularly when coupled with a methanol-soluble diazo initiator: 4,4′-azobis(cyanovaleric acid) (ACVA). Though the choice of methanol as a solvent for a polystyrene-like polymer may seem unusual, it was the experience of the authors that styrene monomer and oligomers are soluble in methanol. Furthermore, the polymers including phosphonium monomers are soluble in methanol at ion content greater than approximately 5 mol%.
The polymerizations were carried out over the course of 24 hours at 60 °C. Using ACVA at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with CTA 1 gives predictable molecular weights as seen in Table 1. Precipitation of the reaction mixture in a solution of 30 vol% isopropanol in n-hexane was followed by a filtration and removal of the residual solvent in a vacuum oven at 75 °C overnight to recover the polymer materials. The samples were further dialyzed against methanol in order to remove the residual monomer. Polymer 3f was polymerized under similar conditions, only this material was synthesized using water as a solvent. The large difference in molecular weight between 3a and 3f is due to the nearly doubling of the repeat unit weight with the addition of the phosphonium chloride functionality as well as the enhanced kinetics of a highly polar system. It was found that highly polar systems polymerize much more quickly over the same period of time. Studies regarding the polymerization kinetics of the ion containing system are ongoing and should reveal reactivity ratios and reaction rates.
:1 ratio with CTA 1 gives predictable molecular weights as seen in Table 1. Precipitation of the reaction mixture in a solution of 30 vol% isopropanol in n-hexane was followed by a filtration and removal of the residual solvent in a vacuum oven at 75 °C overnight to recover the polymer materials. The samples were further dialyzed against methanol in order to remove the residual monomer. Polymer 3f was polymerized under similar conditions, only this material was synthesized using water as a solvent. The large difference in molecular weight between 3a and 3f is due to the nearly doubling of the repeat unit weight with the addition of the phosphonium chloride functionality as well as the enhanced kinetics of a highly polar system. It was found that highly polar systems polymerize much more quickly over the same period of time. Studies regarding the polymerization kinetics of the ion containing system are ongoing and should reveal reactivity ratios and reaction rates.
The properties of these ionomers at elevated temperatures are important, especially for fuel cell applications, where the assembly may be operated at temperatures in excess of 200 °C. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were used to explore the material response to temperature and thermal cycling.
TGA data (Fig. 1a) demonstrate the large effects even minor phosphonium substitutions on the styrenic chain have with hydration ability. The peak associated with the mass loss derivative (Fig. 1b) between 50 and 150 °C corresponds to water loss. There is noticeably more water mass associated with polymer 3f than that of 3a. This is not unreasonable since the high ion content materials are highly water soluble, and will likely carry water with the atmospheric handling of the polymer. The small mass loss around 150–175 °C can be attributed to the decomposition of the residual monomer. The earliest onset of decomposition was observed at 340 °C for the 15 mol% sample, while the 98 mol% sample did not show significant mass loss until almost 400 °C.
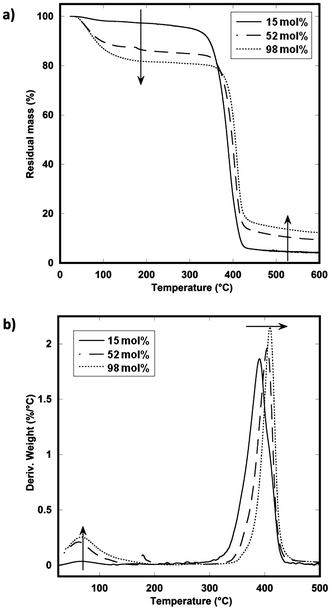 | ||
| Fig. 1 (a) Thermogravimetric mass loss and (b) derivative mass loss of various ion content polystyrenes. | ||
Fig. 2 demonstrates the rise in glass transition temperature (Tg) with increasing ion content in the polymer. These data represent the Tg as measured from the second of three DSC heating cycles at 10 °C min−1 heat/cool rate. Other than the complete loss of water during the first heating cycle, no other transitions were seen between ambient temperatures and 350 °C. Ionomer materials have been seen to demonstrate two Tg transitions: one associated with ion multiplets/clusters and one associated with the nonionic matrix material. While reports from anionic polystyrene ionomers have demonstrated two Tg regions, they tend to be only observable in a small ion content range (1–15 mol% ion)13 and typically only through dynamic mechanical analysis (DMA) testing. Therefore, it would not be expected to see these in this system, nor by this method. The significant rise in Tg is described well by the Gordon, Taylor, and Wood equation for the glass transition behavior of copolymer systems.14,15
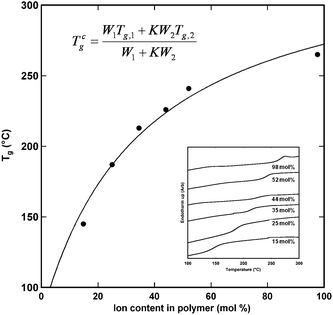 | ||
| Fig. 2 Glass transition temperature as a function of ion content. DSC curves are provided in the inset. | ||
Conclusions
A series of trimethylphosphonium functionalized cationic styrene ionomers was synthesized and thermal properties analyzed. TGA revealed that the copolymers possessed excellent thermal stability. The hydrophilic nature of the high ion content polymers resulted in the incorporation of more atmospheric water than the polymers of lower ionic content. The effect of ion content on the ionomerTg was measured viaDSC, and Tg was found to increase with ion content (copolymer content) following the theory of Gordon, Taylor, and Wood.Acknowledgements
This research was supported in part by the appointment of KKS to the Postgraduate Research Participation Program at the US Army Research Laboratory, administered by the Oak Ridge Institute of Science and Education through an interagency agreement between the US Department of Energy and Army Research Laboratory (contract no. ORISE 1120-1120-99). The identification of commercial equipment and materials in this paper is used to specify adequately the experimental procedures and does not imply recommendations by the Army Research Laboratory.Notes and references
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- L. Carrette, K. A. Friedrich and U. Stimming, ChemPhysChem, 2000, 1, 162–193 CrossRef.
- J. A. Vega and W. E. Mustain, Electrochim. Acta, 2010, 55, 1638–1644 CrossRef CAS.
- A. Tewari, V. Sambhy, M. Urquidi Macdonald and A. Sen, J. Power Sources, 2006, 153, 1–10 CrossRef CAS.
- H. Chen, J.-H. Choi, D. Salas-de la Cruz, K. I. Winey and Y. A. Elabd, Macromolecules, 2009, 42, 4809–4816 CrossRef CAS.
- J. Pan, S. Lu, Y. Li, A. Huang, L. Zhuang and J. Lu, Adv. Funct. Mater., 2010, 20, 312–319 CrossRef CAS.
- S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, Angew. Chem., Int. Ed., 2009, 48, 6499–6502 CrossRef CAS.
- S. R. Williams, W. Wang, K. I. Winey and T. E. Long, Macromolecules, 2008, 41, 9072–9079 CrossRef CAS.
- Handbook of RAFT polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Darmstadt, Germany, 2008 Search PubMed.
- R. Wang and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2468–2483 CrossRef CAS.
- N. Ohtani, T. Yamashita, M. Sugawara and T. Hori, Kobunshi Ronbunshu, 2000, 57, 603–610 CAS.
- N. Ohtani, Y. Inoue, H. Mizuoka and K. Itoh, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2589–2595 CrossRef CAS.
- A. Eisenberg, B. Hird and R. B. Moore, Macromolecules, 1990, 23, 4098–4107 CrossRef CAS.
- M. Gordon and J. S. Taylor, J. Appl. Chem., 1952, 2, 493–500 CAS.
- L. A. Wood, J. Polym. Sci., 1958, 28, 319–330 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and characterization details. See DOI: 10.1039/c0py00293c |
| ‡ Current address: Celgard LLC, Charlotte, NC, USA. |
| This journal is © The Royal Society of Chemistry 2011 |