Synthesis of well-defined multigraft copolymers†
David
Uhrig
a and
Jimmy
Mays
*abc
aCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
bDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA. E-mail: mays@ion.chem.utk.edu
cChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 17th September 2010
Abstract
This short review article focuses on significant developments made in the pursuit of well-defined graft copolymers over the past decade or so. The state-of-the-art for synthesis of the complex copolymers has been greatly advanced over this time period. Anionic polymerization techniques now allow for synthesis of narrow polydispersity multigraft copolymers with control over branch spacing, number of branch points, and branch point functionality. Other controlled/living polymerization techniques are being exploited to increase the chemical diversity of these materials. Future work in this area will focus on synthesis of more complex architectures and incorporation of three or more chemical building blocks into the materials.
 David Uhrig | David Uhrig graduated from the University of Alabama at Birmingham in 2001, earning a PhD in Chemistry under the supervision of Jimmy Mays. He then served a postdoctoral appointment at both the University of South Australia and Flinders University with Professor Janis Matisons. From 2004, he has worked at Oak Ridge National Laboratory, where he currently remains as a technical staff. |
 Jimmy Mays | Jimmy Mays completed his PhD studies at the University of Akron in 1984 under the supervision of Lewis Fetters. Following four years in industry, he joined the Chemistry faculty of the University of Alabama at Birmingham. In 2002 he moved to his present position, a joint appointment at the University of Tennessee and Oak Ridge National Laboratory. His research interests are focused on synthesis of well-defined polymers and copolymers, particularly using living anionic polymerization. |
Introduction
Multigraft copolymers are polymers composed of a main chain of one type of polymer with multiple chemically different side chains connected to the main chain through covalent bonds. Unlike diblock and triblock copolymers, which are linear chains, graft copolymers are a class of branched block copolymers: they have more than two chain ends per molecule. Graft copolymers are important from a scientific perspective because they exhibit properties that reflect the combined effects of thermodynamic incompatibility of the polymer segments and the architectural constraints of the branched architecture. The introduction of a grafted architecture into block copolymers is thus known to influence dynamics,1 bulk morphologies,2,3 and extent of long-range nanoscale order.3,4 The properties of graft copolymers are exploited commercially as adhesives, as emulsifiers and compatibilizing agents, and as tough plastics.5 In the elucidation of structure–property relationships for graft copolymers, progress has been hindered until recently due to synthetic difficulties in producing precisely tailored grafted architectures. Ideally the synthetic chemist would strive to make materials that consist of monodisperse main chains with tethered monodisperse side chains, combined with precise control over the number of branches per molecule, branch point placement, and the number of branches per branch point. All molecules in a synthesized batch of such polymers would thus be exactly the same.Of course, with so many parameters to control, such molecules represent a grand challenge to synthetic polymer chemists and such truly monodisperse and uniform multigraft copolymers have not yet been synthesized. However, great progress has been made toward achieving better control over graft copolymer structure. Various living and controlled polymerizations have been extensively used to produce graft copolymers with narrow molecular weight distribution (MWD) main chains and side chains, with some control over branch number and generally random placement. Living anionic polymerization has thus far yielded the most well-defined multigraft copolymers, which may exhibit very narrow MWD backbones and side chains and with tight control over the number of branch points and branch point placement (see section on Exact control of branch point spacing below). As the desired well-defined materials become architecturally complex, requiring multiple polymerizations and a directed sequence of interdependent manipulations, this control becomes paramount. Unfortunately, only a limited number of monomers are amenable to living anionic polymerization and development of improved multigraft syntheses via other polymerization mechanisms is required. A further and essential criterion in evaluating the merits of a synthetic technique is that the technique must allow for rigorous characterization. Indeed, polymer characterization can oftentimes be as daunting as the synthesis itself. Proof of the claimed architecture can often be established only by thoughtful sampling before, during, and after linking of macromolecular building blocks.
In this review, we focus only on synthesis of well-defined graft copolymers having both narrow MWD backbones and side chains. For synthesis of less well-defined materials, readers are referred to earlier reviews.5,6 For sake of further focus, we are reviewing only materials which have internal polymeric segments between graft points. No effort is made to discuss polymacromonomers, which bear one or more polymeric side chains on each repeating unit in the polymer chain, nor other bottlebrush polymers such as those derived by functionalization of initiation sites to every repeat unit on a polymer backbone. We will begin, in the next section, by reviewing methods that only give tight control over these two molecular parameters. In subsequent sections we discuss strategies that allow control over the number of branches per branch point and branch point spacing. Very recent syntheses of more complex macromolecular architectures incorporating multigraft copolymer components will be summarized, followed finally by conclusions and a summary of prospects for future work in this area.
Conventional synthesis of well-defined multigraft copolymers
Synthesis of graft copolymers of moderately well-defined structures, that is based on narrowly distributed backbone and side chain lengths, using various living and controlled polymerizations was reviewed recently.7 Thus, our intent here is to only review commonly exploited and well-known procedures for making such multigraft copolymers using mostly anionic polymerization.Most syntheses fall into three general strategies: “grafting onto,” “grafting from,” and the “macromonomer” method (also called “grafting through”). In the grafting onto approach both the main and side chains are polymerized before the grafting event, in which functional ends of the side chains are covalently bonded with complementary functionalities along the main chain. The advantage of this strategy lies in the ease of sampling both the main and side chains for characterization. The grafting from approach begins the grafting event with only the main chain pre-polymerized. The main chain next serves as the multi-initiation site from which the side chains are then grown. The advantage of this method is that it is a more expedient synthetic approach. Simply creating reactive sites along the main chain by chemical treatment or irradiation, followed by addition of monomer to generate graft copolymer, makes this an industrially important grafting method. The macromonomer approach employs a preformed side chain that has a polymerizable group at one end of the chain (macromonomer or macromer). Grafting occurs when a regular monomer is polymerized in the presence of the macromonomer, resulting in the main chain being “sewn” through the ends of the side chains in a co-polymerization process. The advantage of this approach is that methods for synthesis of a great variety of macromonomers are well known, and the macromonomer side chains may be thoroughly characterized.
Grafting onto
Adapted to anionic polymerization, the grafting onto method involves the reaction between nucleophilic chain ends of active side chains and electrophilic sites along the main chain. Often the electrophilic sites are introduced after the main chain has been polymerized. One common procedure utilizes chloromethylation of polystyrene (PS).8Chloromethyl groups couple cleanly and quantitatively with oxyanions, but coupling with stronger nucleophiles such as carbanions can often lead to undesirable lithium–halogen exchange reactions. One method of overcoming this side reaction is the conversion of chloromethyl groups into chlorosilyl moieties. By utilizing this strategy, Rahlwes and coworkers9 made well-defined PS-graft-PI (where PI is polyisoprene).Polybutadiene (PBd) can be chlorosilylated through hydrosilation chemistry (Fig. 1).10
 | ||
| Fig. 1 Hydrosilylation of polybutadiene. | ||
Macromolecular carbanions may be reacted directly with the chlorosilyl groups without lithium–halogen exchange. The chlorosilyl group works very well due to the ease of displacement of the Si–Cl bond; this approach is more reliable when the chlorosilyl groups are tethered to the main chain through Si–C bonds since Si–O linkages are themselves labile to powerful nucleophiles. An advantage of these grafting onto strategies is that both the backbone and the side chains may be isolated and independently characterized. Knowing precisely the molecular weights of the building blocks used in constructing the multigraft copolymer helps facilitate its thorough characterization.
Grafting from
Grafting from by anionic polymerization classically employs acid/base chemistry. A main chain may be endowed with a multitude of sites reactive enough to initiate an anionically polymerizable monomer. Acidic hydrogens on amide,11alcohol, or phenol12groups may be removed by tert-BuOK; ethylene oxide (EO) polymerization may then ensue. Hydrogens that are α to a carbonyl group are acidic enough to be removed by lithium diisopropylamide (LDA); the anions thus generated are well suited to the initiation of methacrylic monomers.13With less reactive polymers, anionic sites can be created through metallation of the main chain. Abstraction of more weakly acidic protons (allylic, benzylic, and aromatic) may be accomplished with powerful bases such as sec-BuLi accompanied by chelating compounds such as N,N,N′,N′-tetramethylethylenediamine (TMEDA). TMEDA weakens the C–Li bond through complexation and enhances the basicity of the organolithium compound. In the early 1970s, Falk et al.14 lithiated polydienes by using this method and made a host of poly(diene-g-styrene) materials. When this chemistry was adopted under strict high-vacuum conditions, fractionation of the resulting product provided a fairly well-defined poly(isoprene-g-styrene) (Fig. 2).15
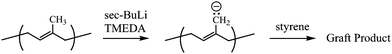 | ||
| Fig. 2 Synthesis of graft copolymers through lithiation of polyisoprene. | ||
An important advance in metallation chemistry, and thus in grafting from using anionic polymerization chemistry, has been the development of what has come to be known as “second generation superbases”.16 In this chemistry, alkyllithiums are enhanced in reactivity through interaction with alkoxides of a less electronegative alkali metal (e.g., a potassium counterion). Due to the favorable association of Li and O, an alkyl-higher alkali metal (e.g., an alkylpotassium compound) is generated in situ. The superbase reagent is capable of quickly deprotonating very modestly acidic substances, where kinetic effects often forbid reactions allowed by thermodynamic stabilities alone. For example, styrenic (aromatic H) and methylstyrenic (benzylic H) residues are rapidly metallated by using this approach. Therefore graft copolymers may be synthesized by the following sequence: (1) polymerization of a suitable monomer (e.g., 4-methylstyrene); (2) metallation; and (3) addition of an anionically polymerizable monomer (Fig. 3).17
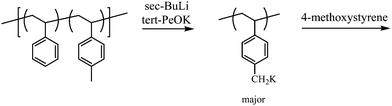 | ||
| Fig. 3 Synthesis of graft copolymers through metallation of poly(styrene-co-4-methylstyrene). | ||
A disadvantage of these grafting from approaches, as compared to grafting onto, is that the side chains may not generally be directly obtained and characterized, although in some cases (i.e.metallation of polydiene backbones) homopolymer of the side chain monomer may be formed during growth of the grafts and may be isolated from the reaction mixture for characterization.
Macromonomer method
Using macromonomers in an anionic grafting polymerization requires addressing the copolymerization behavior of the macromonomer and co-monomer. Disparities in reactivity preferences, expressed by reactivity ratios r1 and r2 of the macromonomer and co-monomer, are generally greater with ionic mechanisms than in free radical polymerization. Thus, it is difficult to control where side chains are placed along the main chain.The most widely used method for the preparation of PS macromonomers is the one developed by Milkovich and Schulz.18 Living polystyryllithium was reacted with ethylene oxide to form the less reactive alkoxide and then with methacryloyl chloride, according to the following scheme (Fig. 4).
 | ||
| Fig. 4 Preparation of polystyrene macromonomer. | ||
Such macromonomers may be copolymerized with ordinary monomers to create multigraft copolymers where branch point spacing is controlled by the reactivity ratios of the two species. When a living or controlled copolymerization mechanism is employed6,7 multigraft copolymers having both narrow polydispersity backbones and branches may be obtained.
However, not much work on anionic copolymerization of macromonomers had been carried out until quite recently because macromonomers are solid materials that are extremely difficult to purify to the standards required for anionic polymerization.19 Recently, it was found20,21 that the anionic homo- or copolymerization of macromonomers leads to persistent anionic species, if the polymerization is performed in situ, i.e. without isolation of macromonomer. The synthesis of the macromonomers involves slow addition of polymer anions to 4-(chlorodimethylsilyl)styrene (CDMSS) (Fig. 5).
 | ||
| Fig. 5 Use of CDMSS to prepare macromonomer, in situ. | ||
The key to the success of this synthesis is that the macromolecular organolithium chain is reacted and consumed more quickly in its substitution reaction with the silyl chloride than in its addition to the styrenic double bond. The selectivity of the macroanion for the Si–Cl over the double bond is greatest for PBLi, then PILi; while PSLi is least selective.22 For cleaner endpoints and graft copolymers, PSLi can be end-capped with a few units of butadiene before titrating into a solution of CDMSS. When the addition reaction starts to take place, noted by formation of a persistent yellow color, initiator, monomer, and a randomizing additive such as potassium alkoxide,23,24 if necessary, is added directly to the macromonomer solution to grow the multigraft copolymer.
Controlling the number of branches per branch point
In the past, nearly all well-defined multigraft copolymers have consisted of a polymer backbone having a single side chain attached to each branch point, which we call trifunctional branch points. Recently, however, strategies have been developed for control of the number of branches per branch point in multigraft copolymers.The grafting onto mechanism has been exploited to create multigraft copolymers carrying two side chains per branch point, i.e. tetrafunctional branch points.25,26 For example, we noted above how hydrosilylation of pendant vinyl groups of polydienes may be employed in a grafting onto strategy by reacting living polymer anions with the resulting pendant chlorosilane functionality. When dichloromethylsilane is employed in the hydrosilylation step, dichlorosilane functionality is introduced at each branching site. These branch points are randomly placed along the polydiene backbone.
The macromonomer approach has also been used to create multigraft copolymers carrying randomly placed tetrafunctional and pentafunctional branch points. This work involved the use of what Hadjichristidis and co-workers termed “double tailed”27 and “triple tailed”28 macromonomers, respectively. This work employed the same in situpolymerization of macromonomers strategy described in the prior section, but instead of CDMSS, 4-(dichloromethylsilyl)styrene (DCMSS) or 4-(2-(dichloromethylsilylethyl)chloromethylsilyl)styrene is used. The synthesis of a PS-g-PBd multigraft having multiple randomly placed tetrafunctional branch points is shown below (Fig. 6).27
 | ||
| Fig. 6 Synthesis of poly(styrene-g-butadiene) through the use of DCMSS and in situ macromonomer preparation. | ||
In an interesting and useful departure from anionic chemistry, Coskun and coworkers29 have prepared double-tailed grafts by acylation of −OH groups in poly[styrene-co-p-bis(2-hydroxyethyl)aminomethyl styrene with α-bromoisobutyryl bromide, and then using this product as a macroinitiator for the atom transfer radical polymerization (ATRP) of isobornyl acrylate. Note that although these products have polydispersity indices (PDIs) of 1.5 and up, that is primarily because the starting backbone material is made from a conventional free radical polymerization of styrene and 4-chloromethylstyrene30 and, in principle, it could be done by a controlled radical polymerization (for example, see ref. 31).
Controlling the number of branches with regular spacing of branch points
Another very different sort of macromonomer strategy has been used to make multigraft copolymers that have regularly spaced tri-, tetra-, and hexa-functional branch points. These materials have been termed “regular combs”, “centipedes” and “barbwire” polymers, respectively. Their synthesis uses chlorosilane coupling chemistry to connect together individual segments of the graft copolymer that bear anions at one chain end or both ends. Chlorosilane linking chemistry has been used in anionic polymerization for nearly 50 years to create model branched polymers such as stars. Starting in 1990,34 this chemistry has been exploited extensively over the past twenty years to create miktoarm stars, as well as other controlled architecture branched block copolymers such as asymmetric miktoarm stars and π and H copolymers.2–4,35The application of this chemistry32,33 to create multigraft copolymers with regularly spaced branch points and tunable branch point functionality makes use of a step growth polymerization mechanism for linking the appropriately functionalized segments together as depicted below (Fig. 7).
 | ||
| Fig. 7 Preparation of multigraft copolymers with regularly spaced branch points through a polycondensation approach. | ||
The polydispersity indices of the resulting centipede copolymers are just over 2, as expected for a step growth mechanism, and the average number of branch points can be controlled by reaction stoichiometry (Carothers equation). Large numbers of branch points (>10) and very high molecular weights (hundreds of thousands) may be achieved for combs, centipedes, and barbwires with careful control of the chemistry and stoichiometry.33
While the broad molecular weight distribution of the products might appear to be a major disadvantage of this technique, it is important to realize that the polydispersity reflects almost exclusively multigraft copolymers having different numbers of branch points. Fractionation of the raw products yielded specimens of narrow polydispersities (PDIs of about 1.2) which are composed of exactly the same side chains and backbone units but which differ only in number of branch points. Such specimens are ideal materials for studying the influence of number of branch points on copolymer properties, and have been extensively explored in probing morpholgy,36 dynamics,1 and mechanical properties37 of multigraft copolymers. Particularly noteworthy was the finding that certain compositions of regular multigraft copolymers exhibited superelasticity, unprecedented elongation at break and very low hysteresis, with both tensile strength and strain at break increasing with number of branch points for a given type of material.37
Plamper and coworkers have recently prepared multigraft copolymers having a polyethylene oxide (PEO) backbone and poly(dimethylaminoethyl methacrylate) (PDMAEMA) grafts.38 The PEO backbone was prepared by the polycondensation of mesylated dihydroxy-telechelic PEO with a low molecular weight polyalcohol, dipentaerythritol (a polyol with 6 –OH groups), 4 of the –OHs protected. After deprotection of the hydroxyls, PDMAEMA was grafted by either transforming the –OH groups to ATRP initiation sites and adding monomer, or by transforming the –OHs to alkynes and reacting them with premade azide-functionalized PDMAEMA. The PDMAEMA grafts were clustered into groups of 4, and these clusters were controllably spaced along the PEO backbone, according to the MW of the dihydroxy-telechelic PEO starting material.
Exact control of branch point spacing
As seen in the prior section, regular spacing of branch points and control of number of branches per branch point has been achieved, although in some instances fractionation is required to obtain materials exhibiting uniform backbone lengths and number of branch points. Work by several groups has been carried out with the goal of achieving the synthesis of the ideal graft copolymer synthesis outlined in the Introduction: uniform side chains connected to a uniform backbone at precisely chosen locations along the backbone with the same number of branches on every molecule.One strategy that enhances the utility of the macromonomer approach in anionic polymerization is the use of 1,1-diphenylethylene (DPE)—end-capped macromonomers.39 Since DPE does not homopolymerize, monomer and macromonomer can be added in an alternating manner (Fig. 8).
 | ||
| Fig. 8 Preparation of graft copolymers through a DPE-based strategy. | ||
The synthetic advantage of DPE anions avoids the usual scenario for macromonomer-based synthesis of graft copolymers: dependence on reactivity ratios of the macromonomer and regular monomer in governing branch point placement. The DPE technique, with careful control of stoichiometry, actually allows placement of the side chain of desired molecular weight and composition at a predetermined location along the main chain.
Recently comb-shaped graft copolymers have been synthesized wherein all molecular parameters have been tightly controlled: backbone molecular weight, graft molecular weight, graft placement and, particularly, distinct control of each graft attachment in terms of its uniquely decided molecular weight and placement. These materials have been aptly termed “exact” graft copolymers. Paraskeva and Hadjichristidis40 synthesized exact graft copolymers by making use of 1,4-bis-(1-phenylethenyl)benzene (1,4-PEB) and its ability to intercept one carbanion at a time (due to mesomeric repulsion of a second incoming carbanion until the first adducted anion is neutralized). 1,4-PEB was used to intercept the carbanionically active end of the backbone; after neutralizing and work-up, a carbanionically active graft was appended to the PEB-functionalized backbone, and the resultant carbanion adduct was used as the initiation moiety for a fresh portion of monomer to continue the backbone further. This sequence is repeatable for multiple graft attachments. Hirao and co-workers41,42 have made exact graft copolymers by another “double-DPE” approach. A segment of α-TBDMSiO-protected, ω-anionically active backbone was incorporated to 1,4-PEB and the resultant anion was adducted to another segment of backbone bearing a primary alkyl bromide end-group. This neutralized in-chain PEB was next able to capture an anionically active graft chain. The graft copolymer product was worked up, and the TBDMSiO group was deprotected and transformed into another α-TBDMSiO group. Hirao and co-workers have demonstrated the repeatability of this sequence for preparation of a copolymer with 5 grafts. Both of these exact graft chemistries are admirable for that they lead to complex graft copolymers of highly tunable structure, with narrow MWD side chains and backbones, control over placement of side chains along the backbone, and the potential to employ side chains having different molecular weights and compositions. Limitations of the chemistries are that they require laborious multistep syntheses and they are therefore limited, practically, to relatively small numbers of branches and modest molecular weights. (For example, for a multigraft copolymer to function as a strong thermoplastic elastomer, high molecular weight segments are desired.37) Control of branch point functionality beyond trifunctional has not yet been demonstrated but should be possible.
Complex macromolecular architectures incorporating multigraft copolymer segments
Koutalas et al.43 prepared well-defined graft-block-graft copolymers through the use of styrenic macromonomers, derived from chlorodimethylsilylstyrene-capped macroanions. The synthetic approach involves the in situcopolymerization of these macromonomers in the presence of monomer and trace potassium alkoxide randomizer. By sequentially polymerizing differing complements of monomer and macromonomer, complicated yet still very well defined multigraft copolymers were made (Fig. 9). | ||
| Fig. 9 Complex multigraft copolymers. | ||
By using a clever combination of nitroxide-mediated radical polymerization (NMP), anionic polymerization, and DPE chemistry, Yang and coworkers44 have recently synthesized multigrafts with not only “V-shaped” (or “double-tailed” or “centipede” depending upon choice of nomenclature) grafts but also “Y-shaped” grafts. A polystyrene backbone was prepared by NMP copolymerization of styrene and 4-chloromethylstyrene. Grafts were prepared by sequentially incorporating two macroanions into 1,4-PEB, and the resulting anions were either grafted to the backbone directly, for V-shaped multigrafts, or used to initiated the polymerization of styrene or isoprene before lastly reacting with the backbone, yielding Y-shaped multigrafts (Fig. 10).
 | ||
| Fig. 10 Multigraft copolymers obtained through a combination of NMP, anionic, and DPE chemistry. | ||
Yang and coworkers have also prepared graft-on-graft multigrafts (see Fig. 11a) wherein the same type of poly(styrene-co-4-chloromethylstyrene) was first grafted onto, simply, by polyisoprenyl anions. These PI grafts were epoxidized and the epoxy groups were grafted onto by polystyryl anions.31
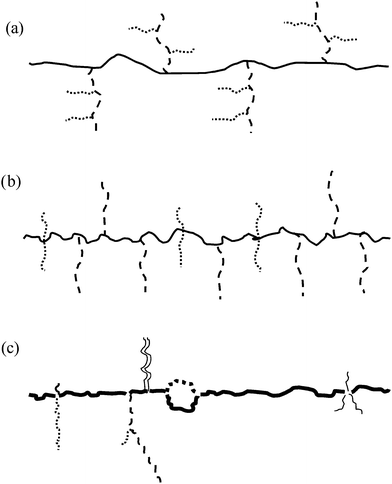 | ||
| Fig. 11 Future multigraft architectures: (a) graft on graft; (b) mixed multigraft; (c) complex exact graft. | ||
Conclusions and future prospects
Development of chemistry to synthesize tailored and well-defined multigraft copolymers has made remarkable progress over the past decade. The standard for a “model multigraft copolymer” is no longer limited to controlling the MW and MWD of the backbone and side chains. Precision control over branch point placement and branch point functionality is now possible. In addition, we have already seen the synthesis of complex and beautiful architectures such as graft-block-graft and graft-on-graft copolymers. There are limitless possibilities for synthesis of increasingly complex grafted architectures (for examples, see Fig. 11). Some general future developments that we anticipate include:• Exploiting three or more monomers to create complex multifunctional architectures, exploiting a general trend in block copolymers, of which multigraft copolymers are nonlinear analogues. This work has already begun, as noted in the previous section. The exact graft copolymer synthesis strategy is certain to be exploited for this purpose, as every branch introduced via this approach can be chemically different.
• Advances in controlled polymerization methods besides anionic polymerization. Anionic polymerization is clearly the method at present that leads to the most well-defined multigraft copolymers. However, it is limited to only a few monomers. Advances in multigraft synthesis via other controlled polymerization mechanisms will allow a wide range of functional multigraft copolymers to be synthesized, expanding the range of their potential applications.
• Use of self-assembly in creating reversible multigraft copolymers, perhaps in response to an external stimulus. Polypeptide-based materials may play a key role here.
• Advances in polymer characterization methods such as temperature gradient interaction chromatography (TGIC) will “raise the bar” for synthesis of tailored multigraft copolymers (and for all tailored polymer synthesis!). TGIC is an emerging polymer characterization technique with vastly superior resolution to size exclusion chromatography (SEC) and with far less band broadening.45 It can, for example, resolve low levels of impurities in model copolymers that are not readily detected by SEC.46 A diblock copolymer synthesized by living anionic polymerization and exhibiting an extremely narrow MWD by SEC has been fractionated by TGIC to obtain dozens of fractions exhibiting different composition, MWs, and morphologies.47
Acknowledgements
This review work was supported by the Center for Nanophase Materials Sciences, which is sponsored at Oak Ridge National Laboratory by the Division of Scientific User Facilities, United States Department of Energy.References
- J. Mijovic, M. Sun, S. Pejanovic and J. W. Mays, Macromolecules, 2003, 36, 7640 CrossRef CAS.
- S. T. Milner, Macromolecules, 1994, 27, 2333 CrossRef CAS; N. Hadjichristidis, H. Iatrou, S. K. Behal, J. J. Chludzinski, M. M. Disko, R. T. Garner, K. S. Liang, D. J. Lohse and S. T. Milner, Macromolecules, 1993, 26, 5812 CrossRef CAS.
- D. J. Pochan, S. P. Gido, S. Pispas, J. W. Mays, A. J. Ryan, P. Fairclough, N. Terrill and I. W. Hamley, Macromolecules, 1996, 29, 5091 CrossRef CAS; C. Lee, S. P. Gido, M. Pitsikalis, J. W. Mays, N. Beck Tan, S. Trevino and N. Hadjichristidis, Macromolecules, 1997, 30, 3732 CrossRef CAS.
- F. L. Beyer, S. P. Gido, C. Buschl, H. Iatrou, D. Uhrig, J. W. Mays, M. Y. Chang, B. A. Garetz, N. P. Balsara, N. B. Tan and N. Hadjichristidis, Macromolecules, 2000, 33, 2039 CrossRef CAS; J. W. Mays, D. Uhrig, S. Gido, Y. Zhu, R. Weidisch, H. Iatrou, N. Hadjichristidis, K. Hong, F. Beyer, R. Lach and M. Buschnakowski, Macromol. Symp., 2004, 215, 111 CrossRef CAS.
- J. M. G. Cowie, Block and Graft Copolymers, in Comprehensive Polymer Science, ed. G. Allen and J. C. Bevington, Pergamon, Oxford, 1989, vol. 3, p. 33 Search PubMed; P. Dreyfus and R. P. Quirk, Graft Copolymers, in Encyclopedia of Polymer Science and Engineering, ed. J. I. Kroshwitz, Wiley-Interscience, New York, 2nd edn, 1987, vol. 7, p. 551 Search PubMed; N. Hadjichristidis, M. Pitsikalis, H. Iatrou, P. Driva, M. Chatzichristi, and G. Sakellariou, Graft Copolymers, in Encyclopedia of Polymer Science and Technology, ed. A. Seidel, John Wiley, New York, 2010, in press Search PubMed.
- M. Pitsikalis, S. Pispas, J. W. Mays and N. Hadjichristidis, Adv. Polym. Sci., 1998, 135, 1 CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS.
- T. Altares, Jr, D. P. Wyman, V. R. Allen and K. J. Meyersen, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 4131 CrossRef; F. Candau and P. Rempp, Makromol. Chem., 1969, 122, 15 CrossRef CAS; K. W. Pepper, H. M. Paisley and M. A. Young, J. Chem. Soc., 1953, 4097 RSC; S. Itsuno, K. Uchikoshi and K. Ito, J. Am. Chem. Soc., 1990, 112, 8187 CrossRef CAS.
- D. Rahlwes, J. E. L. Roovers and S. Bywater, Macromolecules, 1977, 10, 604 CrossRef CAS.
- G. C. Cameron and M. Y. Qureshi, Makromol. Chem. Rapid Commun., 1981, 2, 287 CrossRef CAS.
- P. Jannasch and B. Wesslen, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1465 CrossRef CAS.
- K. Se, O. Watanabe, T. Shibamoto and T. Fujimoto, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1988, 29(2), 110 CAS.
- M. Inoki, F. Akutsu, H. Yamaguchi, K. Naruchi and M. Miura, Macromol. Chem. Phys., 1994, 195, 2799 CrossRef CAS.
- J. C. Falk, R. J. Scott, D. F. Hoeg and F. Pendleton, Rubber Chem. Technol., 1973, 46, 1044 CAS.
- N. Hadjichristidis and J. Roovers, J. Polym. Sci., Part A: Gen. Pap., 1978, 16, 851 Search PubMed.
- L. Lochmann and J. M. J. Frechet, Macromolecules, 1996, 29, 1767 CrossRef CAS; J. H. G. Steinke, S. A. Haque, J. M. J. Frechet and H. C. Wang, Macromolecules, 1996, 29, 6081 CrossRef CAS.
- M. Janata, L. Lochmann, J. Brus, P. Holler, Z. Tuzar and P. Kratochvil, Macromolecules, 1997, 30, 7370 CrossRef CAS; B. D. Edgecombe, J. A. Stein, J. M. J. Frechet, Z. Xu and E. J. Kramer, Macromolecules, 1998, 31, 1292 CrossRef CAS.
- G. O. Schulz and R. Milkovich, J. Appl. Polym. Sci., 1982, 27, 4773 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, S. Pispas and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3211 CrossRef CAS; D. Uhrig and J. W. Mays, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6179 CrossRef CAS.
- D. M. Knauss, A. H. Al-Muallem, T. Huang and D. T. Wu, Macromolecules, 2000, 33, 3557 CrossRef CAS; H. A. Al-Muallem and D. M. Knauss, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3547 CrossRef CAS.
- D. Pantazis, I. Chalari and N. Hadjichristidis, Macromolecules, 2003, 36, 3783 CrossRef CAS.
- A. Vazaios and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1038 CrossRef CAS.
- C. F. Wofford and H. L. Hsieh, J. Polym. Sci., Part A: Gen. Pap., 1969, 7, 461 Search PubMed.
- G. Koutalas, H. Iatrou, D. Lohse and N. Hadjichristidis, Macromolecules, 2005, 38, 4996 CrossRef CAS.
- M. Xenidou and N. Hadjichristidis, Macromolecules, 1998, 31, 5690 CrossRef CAS; G. Velis and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1136 CrossRef CAS.
- A. Hirao and S. W. Ryu, Macromol. Symp., 2003, 192, 31 CrossRef CAS.
- P. Driva, H. Iatrou, D. J. Lohse and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4070 CrossRef CAS.
- A. Nikopoulou, H. Iatrou, D. J. Lohse and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3513 CrossRef CAS.
- P. Seven, M. Coskun and K. Demirelli, React. Funct. Polym., 2008, 68, 922 CrossRef CAS.
- M. Coskun and M. M. Temuz, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 668 CrossRef CAS.
- W. Sun, F. Yu, J. He, C. Zhang and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5518 CrossRef CAS.
- H. Iatrou, J. W. Mays and N. Hadjichristidis, Macromolecules, 1998, 31, 6697 CrossRef CAS.
- D. Uhrig and J. W. Mays, Macromolecules, 2002, 35, 7182 CrossRef CAS.
- J. W. Mays, Polym. Bull., 1990, 23, 247 CrossRef CAS.
- H. Iatrou, E. Siakali-Kioulafa, N. Hadjichristidis, J. Roovers and J. W. Mays, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1925 CAS; D. J. Pochan, S. P. Gido, S. Pispas and J. W. Mays, Macromolecules, 1996, 29, 5099 CrossRef CAS; S. Pispas, J. W. Mays, D. Pochan, S. P. Gido and N. Hadjichristidis, Macromolecules, 1996, 29, 7022 CrossRef CAS; C. Lee, S. P. Gido, Y. Poulos, N. Hadjichristidis, N. Beck Tan, S. F. Trevino and J. W. Mays, J. Chem. Phys., 1997, 107, 6460 CrossRef CAS; C. Lee, S. P. Gido, M. Pitsikalis, J. W. Mays, N. Beck Tan, S. Trevino and N. Hadjichristidis, Macromolecules, 1997, 30, 3732 CrossRef CAS; C. Lee, S. P. Gido, Y. Poulos, N. Hadjichristidis, N. B. Tan, S. F. Trevino and J. W. Mays, Polymer, 1998, 39, 4631 CrossRef CAS.
- F. L. Beyer, S. P. Gido, C. Buschl, H. Iatrou, D. Uhrig, J. W. Mays, M. Y. Chang, B. A. Garetz, N. P. Balsara, N. B. Tan and N. Hadjichristidis, Macromolecules, 2000, 33, 2039 CrossRef CAS.
- R. Weidisch, S. P. Gido, D. Uhrig, H. Iatrou and J. Mays, Macromolecules, 2001, 34, 6333 CrossRef CAS; J. W. Mays, D. Uhrig, S. Gido, Y. Zhu, R. Weidisch, H. Iatrou, N. Hadjichristidis, K. Hong, F. Beyer, R. Lach and M. Buschnakowski, Macromol. Symp., 2004, 215, 111 CrossRef CAS; U. Staudinger, R. Weidisch, Y. Zhu, S. P. Gido, D. Uhrig, J. W. Mays, H. Iatrou and N. Hadjichristidis, Macromol. Symp., 2006, 233, 42 CrossRef CAS; Y. Zhu, E. Burgaz, S. P. Gido, U. Staudinger, R. Weidisch, D. Uhrig and J. W. Mays, Macromolecules, 2006, 39, 4428 CrossRef CAS; Y. Duan, E. Rettler, K. Schneider, R. Schlegal, M. Thunga, R. Weidisch, H. W. Siesler, M. Stamm, J. W. Mays and N. Hadjichristidis, Macromolecules, 2008, 41, 4565 CrossRef CAS; U. Staudinger, R. Schlegel, R. Weidisch, J. Fritzsche, M. Kluppel, G. Heinrich and J. W. Mays, Eur. Polym. J., 2008, 44, 3790 CrossRef CAS; M. Thunga, R. Schlegel, U. Staudinger, Y. Duan, R. Weidisch, G. Heinrich, J. Mays and N. Hadjichristidis, KGK, Kautsch. Gummi Kunstst., 2008, 61, 597 Search PubMed; Y. Duan, M. Thunga, R. Schlegel, K. Schneider, E. Rettler, R. Weidisch, H. Siesler, M. Stamm, J. W. Mays and N. Hadjichristidis, Macromolecules, 2009, 42, 4155 CrossRef CAS; R. Schlegel, D. Wilkin, Y. Duan, R. Weidisch, G. Heinrich, D. Uhrig, J. W. Mays and N. Hadjichristidis, Polymer, 2009, 50, 6297 CrossRef CAS; R. Schlegel, U. Staudinger, M. Thunga, R. Weidisch, G. Heinrich, D. Uhrig, J. W. Mays, H. Iatrou and N. Hadjichristidis, Eur. Polym. J., 2009, 45, 2902 CrossRef CAS.
- F. Plamper, S. Reinicke, M. Elomaa, H. Schmalz and H. Tenhu, Macromolecules, 2010, 43, 2190 CrossRef CAS.
- T. Fujimoto, H. Zhang, T. Kazama, Y. Isono, H. Hasegawa and T. Hashimoto, Polymer, 1992, 33, 2208 CrossRef CAS.
- S. Paraskeva and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 931 CrossRef CAS.
- A. Hirao, T. Watanabe and R. Kurokawa, Macromolecules, 2009, 42, 3973 CrossRef CAS.
- A. Hirao, K. Murano, R. Kurokawa, T. Watanabe and K. Sugiyama, Macromolecules, 2009, 42, 7820 CrossRef CAS.
- G. Koutalas, D. Lohse and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4040 CrossRef CAS.
- F. Yu, J. He, X. Wang, G. Gao and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4013 CrossRef CAS.
- T. Chang, Adv. Polym. Sci., 2003, 163, 1 CAS.
- S. Park, D. Cho, K. Im, T. Chang, D. Uhrig and J. Mays, Macromolecules, 2003, 36, 5834 CrossRef CAS.
- S. Park, K. Kwon, D. Cho, B. Lee, M. Ree and T. Chang, Macromolecules, 2003, 36, 4662 CrossRef CAS.
Footnote |
† νũν ![[small alpha, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e14c.gif) πολύεις τóν δοũλόν σου, δέσποτα, κατ τò πολύεις τóν δοũλόν σου, δέσποτα, κατ τò ![[small rho, Greek, dot above]](https://www.rsc.org/images/entities/char_e0a6.gif) ![[small eta, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e110.gif) μά σου έν εíρήνη (Lord, you are now dismissing your servant in peace). We dedicate this article to our friend and collaborator Professor Nikos Hadjichristidis on the occasion of his retirement. Nikos has made many pioneering contributions to the field of multigraft copolymers, as documented in this article. We sincerely doubt retirement will reduce Nikos' continuing scientific impact, so we expect to see much further great work. μά σου έν εíρήνη (Lord, you are now dismissing your servant in peace). We dedicate this article to our friend and collaborator Professor Nikos Hadjichristidis on the occasion of his retirement. Nikos has made many pioneering contributions to the field of multigraft copolymers, as documented in this article. We sincerely doubt retirement will reduce Nikos' continuing scientific impact, so we expect to see much further great work. |
| This journal is © The Royal Society of Chemistry 2011 |