Electrodeposition and properties of TEMPO functionalized polythiophene thin films†
Tamara K.
Kunz
and
Michael O.
Wolf
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: mwolf@chem.ubc.ca; Fax: +1-604-822-2847; Tel: +1-604-822-1702
First published on 25th November 2010
Abstract
The modification of an azide-functionalized regioregular polythiophene with TEMPO groups is reported. The TEMPO functionalized polymer is characterized by ESR, ATR-IR, absorption and emission spectroscopies and electrochemical methods. Partial quenching of the polymer emission by the TEMPO groups is observed. The polymer may be electrodeposited onto ITO or carbon electrodes to give thin films. Cyclic voltammetry gives specific charge capacity values of 2.3 mA h g−1 for the TEMPO functionalized polymers, and specific capacitances of 0.8 F g−1 determined by galvanostatic charge/discharge experiments. The reduced charge capacity relative to unfunctionalized polythiophene is attributed to TEMPO induced cleavage and oxidation of the conjugated backbone.
Introduction
Organic materials capable of reversibly storing charge are of significant interest for use in devices such as batteries1 or supercapacitors.2 These materials offer several key advantages over traditional inorganic battery materials. They are low weight, offer mechanical flexibility and are compatible with solution based processing techniques such as spin-coating3 and ink-jet printing.4Conjugated polymers (CPs) have been investigated as potential battery materials5 as they possess many desirable characteristics for this application: they can be reversibly doped, can be prepared in thin-film form, and can be made soluble via substituents on the polymer backbone. However, CPs often exhibit low doping levels, resulting in small redox capacities, and slow kinetics at the electrode often limit the charging and discharging rates of the resulting device.6An alternative approach involves the use of redox polymers where discrete moieties, which undergo reversible redox processes, are attached to a polymer backbone and can be used to store charge. Promising candidates for charge storage are stable radical species that have electrochemically reversible redox properties.6 A neutral radical (R˙) can be oxidized to a cation (R+) or reduced to an anion (R−). Electrochemical reversibility is dependent on the stability of the parent R˙, and the closed shell products R+ and R−. Nitroxide radicals, contain a ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) N–O group upon which the unpaired electron spin density is located. A key feature for the stability of the nitroxide radical 2,2,6,6-tetramethylpiperindine-1-oxyl (TEMPO) is the absence of α-hydrogen atoms. TEMPO has found widespread applications as a catalyst for the oxidation of alcohols,7 as a biological radical scavenger,8 and as a mediator in living free radical polymerizations.9TEMPO has also been used as a pendant group on polynorbornene polymers.1 Due to its rapid and stoichiometric reversible redox processes,10 it offers high-charging and discharging rates, making it potentially useful in battery applications.
N–O group upon which the unpaired electron spin density is located. A key feature for the stability of the nitroxide radical 2,2,6,6-tetramethylpiperindine-1-oxyl (TEMPO) is the absence of α-hydrogen atoms. TEMPO has found widespread applications as a catalyst for the oxidation of alcohols,7 as a biological radical scavenger,8 and as a mediator in living free radical polymerizations.9TEMPO has also been used as a pendant group on polynorbornene polymers.1 Due to its rapid and stoichiometric reversible redox processes,10 it offers high-charging and discharging rates, making it potentially useful in battery applications.
Here, a new polythiophene (PT) functionalized with pendant TEMPO radicals is described. This material can take advantage of the redox capabilities of both the TEMPO and PT backbone to potentially increase the charge stored per unit weight. Investigations of this potential charge storage capacity are presented and discussed.
Experimental
General
All reagents and solvents were used as received except where noted. Tetrahydrofuran (THF) was distilled from Na/benzophenone. Anhydrous dimethylformamide (DMF) was distilled from P2O5. Compound 27b and poly-111 were prepared according to the literature procedures.1H NMR spectra were obtained in CDCl3 either on a Bruker AV-400 Inverse or a Bruker AV-400 Direct spectrometer. Chemical shifts are reported in ppm, referenced to residual chloroform (CHCl3) (δ 7.27). ATR-IR spectra were recorded on a Thermo Scientific Nicolet 6700 FT-IR spectrometer as neat powders or oils. Electronic absorption spectra were obtained on a Varian Cary 5000 UV-vis spectrometer in HPLC grade CH2Cl2 or CHCl3. Fluorescence measurements were made on a PTI Quantamaster spectrometer in the same solvents used for UV-vis measurements. Gel permeation chromatography (GPC) was performed using a Waters GPC equipped with 3 µg Styragel columns against polystyrene standards. The polymer was dissolved, filtered and eluted with THF at a flow rate of 1.0 mL min−1 and monitored with a UV-vis detector (Waters 2487). Electrochemical measurements were conducted on an Autolab PGSTAT12 using either a Pt disk, ITO plated glass or a microporous carbon paper working electrode, Pt mesh counter electrode, and a Ag wire reference electrode. These measurements were made in either CH2Cl2 passed through an alumina column, acetonitrile distilled over molecular sieves (type 3A, 4–8 mesh beads) or propylene carbonate obtained from Aldrich. Unless specified, measurements were referenced against a pseudo-reference Ag wire. Alternatively, an internal reference (decamethylferrocene) was added to correct the measured potentials with respect to the saturated calomel electrode (SCE). The 0.1 M [n-Bu4N]PF6 supporting electrolyte was purified by triple recrystallization from ethanol and dried at 90 °C under vacuum for 3 days. Electron spin resonance (ESR) spectra were obtained using a Bruker Elexys E-500, X-band (9.5 GHz) spectrometer. Solution samples were measured in benzene solution.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500, Mn = 8500, PDI = 1.5.
500, Mn = 8500, PDI = 1.5.
Results and discussion
Synthesis and characterization
The modification of a regioregular polymer (prepared by Grignard metathesis (GRIM)12polymerization) via attachment of a stable free radical was carried out to investigate the potential charge storage capabilities of a coupled organic redox group and a CP. Poly-3 was synthesized using Click chemistry to modify the azide functionalized copolymer, poly-1,11 with an acetylene modified TEMPO radical (2) as shown in Scheme 1. Poly-1 was designed to be soluble in organic solvents due to the hexyl side chains, and with sufficient distance between the reactive azide groups on the backbone to avoid excessive steric interactions with bulky substituents. Previously,11 Click reactions with poly-1 were carried out using CuSO4·5H2O and L-ascorbic acid to generate the active catalystin situ. It has been demonstrated that nitroxyls of the piperidine series can be oxidized by CuII in solution.13 To avoid any complications with the reactivity of the catalyst, CuI was used in place of CuSO4·5H2O and ascorbic acid.7b,14Poly-3 was isolated and purified via multiple re-precipitations from methanol.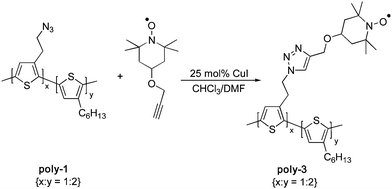 | ||
| Scheme 1 Synthesis of poly-3. | ||
Poly-3 was characterized by ATR-IR, UV-vis, fluorescence, 1H NMR, and ESR spectroscopies. In the ATR-IR spectrum, the azide stretching band of poly-1 at 2091 cm−1 disappears. In addition, there are no acetylene stretches present at 3229 or 2111 cm−1. This confirms that the azide and acetylene starting materials have reacted. TEMPO lacks a distinct IR handle for confirmation of its presence; thus closer inspection of the fingerprint region of the IR spectra was necessary (Fig. S1†). A band observed at 1361 cm−1 is within the characteristic range of the NO˙ moiety (1340–1390 cm−1)15 and supports the presence of the free radical. Further support of this assignment is obtained from a band at 1365 cm−1 observed for the TEMPO precursor, 2, and the lack thereof in poly-1.
Absorbance and emission studies were carried out in CH2Cl2 solution (Fig. 1). The absorbance and emission maxima of poly-1 and poly-3 are identical, at 460 and 575 nm respectively. The absorbance and emission of both polymers are due to the π–π* transition localized on the conjugated backbone. The non-conjugated ethyl linker between the backbone and the azide in poly-1 and the TEMPO group in poly-3 prevents any inductive interactions which could cause shifts in the electronic spectra between the two polymers. Interestingly, there is a significant reduction in fluorescence intensity for poly-3 compared to that of poly-1 when the two compounds were studied in CH2Cl2 solutions of the same optical density at the excitation wavelength. Oxidative quenching of poly-3 by the pendant TEMPO group may be the cause of this decrease. The TEMPO group is commonly used as a one electron acceptor in fluorescence quenching experiments.16 The incomplete fluorescence quenching is attributed to a concentration of TEMPO groups that is too low to fully quench the system.
 | ||
| Fig. 1 Absorption and emission spectra of CH2Cl2 solutions of poly-3 (red) and poly-1 (black). | ||
The 1H NMR spectrum of poly-3 shows broadened signals, as expected for a polymeric radical-containing material.17 The proton resonances of the two methylene groups of the ethyl azide chain in poly-1 appear at δ 3.60 and 3.12. The attachment of the TEMPO radical makes these protons difficult to resolve in the spectrum of poly-3 due to the proximity of the free radical. The signals from δ 2.80 to 0.91 are assigned to the protons of the hexyl groups and correspond well in chemical shift with corresponding resonances in poly-1.
Electron spin resonance
Systems that contain one or more unpaired electrons result in line broadening in NMR spectroscopy.17 Electron spin resonance (ESR) spectroscopy was carried out on poly-3 to further confirm the presence of the TEMPO moiety (Fig. S2†). The ESR spectrum of poly-3 in a benzene solution at room temperature shows a triplet centered at 3504 G, characteristic of TEMPO derivatives, confirming that the radical is present.18Cyclic voltammetry
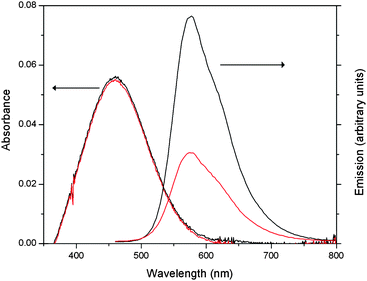
Poly-3 was studied using cyclic voltammetry (CV) using indium tin oxide (ITO) on glass and high surface area microporous carbon paper working electrodes. In the studies of poly-3 carried out on ITO coated glass slides (Fig. 2a), the first scan of the voltammogram in CH2Cl2 displays an oxidation wave at 1.28 V (vs.Ag wire) attributed to oxidation of TEMPO with the corresponding reduction wave observed at 1.06 V (vs.Ag wire). Interestingly, sequential scans exhibit increasing current, with a noticeable shift of the TEMPO redox wave to higher potential. This behaviour is characteristic of conducting polymer deposition on an electrode and is typically observed in monomers which polymerize electrochemically.![Cyclic voltammograms of the growth of (a) poly-3 and (b) poly-1. ITO coated glass electrodes in 0.1 M [n-Bu4N]PF6 in CH2Cl2. Scan rate = 50 mV s−1.](/image/article/2011/PY/c0py00308e/c0py00308e-f2.gif) | ||
| Fig. 2 Cyclic voltammograms of the growth of (a) poly-3 and (b) poly-1. ITO coated glass electrodes in 0.1 M [n-Bu4N]PF6 in CH2Cl2. Scan rate = 50 mV s−1. | ||
To our knowledge, this type of electrochemical deposition has not been previously reported for preformed CPs. The same type of electrodeposition was also observed for poly-1 (Fig. 2b), demonstrating that this is not a consequence of inclusion of the TEMPO moiety in the polymer. The redox wave of the TEMPO group is superimposed on the redox features of the polymer as it electrodeposits. Masses of deposited polymer on carbon paper were typically between 0.5 and 1.2 mg.
After electrodeposition on ITO, the film was removed from the polymerization solution, washed with CH2Cl2 to remove any extraneous, undeposited polymer, and the cyclic voltammetry subsequently studied in polymer-free solution. In CH2Cl2, upon applying a potential scan (−0.2 V to 1.8 V) to the thin film electrode, no current was recorded. In CH3CN solution, a separate film was examined over the same potential range and the current was observed to decrease with sequential scans (Fig. S3†). During this experiment there was the appearance of a purple halo-like cloud around the electrode suggesting that the polymer dissolves from the electrode during electrochemical cycling.
Similar redox features and film instability were observed for the thin films of poly-3 deposited on carbon paper electrodes in both CH2Cl2 and CH3CN. The oxidation and reduction waves of the TEMPO groups were observed at 0.91 V and 0.85 V (vs.SCE) on carbon paper, respectively. The carbon paper electrodes were also studied in a propylene carbonate solution, where the deposited polymer films of poly-3 were found to be more stable under electrochemical cycling, allowing additional measurements to be conducted. Fig. 3 shows the cyclic voltammograms of a thin film of poly-3 on a carbon paper electrode at various scan rates. Peak current increases linearly with scan rate demonstrating reversible redox behaviour and rapid electron transfer kinetics.19
 | ||
| Fig. 3 Cyclic voltammograms of poly-3 on carbon paper in propylene carbonate at 25, 50, 100, 150, and 200 mV s−1. | ||
Capacity and charge/discharge studies
The films of 1 deposited on carbon paper electrodes were used to measure the charge capacity of the polymer films. Integration of the CV curve over the oxidation scan from 0.0 to 1.5 V yields a measure of the amount of charge that can be stored in the material over that potential range. The value determined by integration of the CV curves is 2.3 mA h g−1 (averaged over several samples.) This was similar in magnitude to the capacity determined for films of poly-1 on carbon paper prepared in the same manner. In comparison, polythiophene-based battery cells have been shown to have specific charge capacities ranging from 24–98 mA h g−1 depending on conditions,5 thus poly-3 and poly-1 both perform more poorly than unsubstituted PT.Further evaluation of the electrochemical behavior of the polymer films was done by carrying out galvanostatic charge and discharge cycles. An electrodeposited film was placed in a polymer-free solution and with a constant applied current the potential was scanned in both positive and negative directions within the potential range of charge storage. Fig. 4 shows the charge/discharge curves from scans at 0.01 mA, cycled between 0.8 and 1.1 V for poly-3. The specific capacitances of these films on the first cycle were calculated to be ∼0.8 F g−1. Charge and discharge cycles were also carried out on poly-1, and delivered values an order of magnitude higher at 11.3 F g−1. Multiple samples were tested, and poly-3 consistently displayed lower specific capacity values than poly-1. In comparison, the specific capacitance of chemically polymerized polythiophene is 260 F g−1,20 much greater than the values obtained for either poly-1 or poly-3.
![Typical galvanostatic charge–discharge curve of poly-3 scanned at 0.01 mA in propylene carbonate with 0.1 M [n-Bu4N]PF6 supporting electrolyte. Scanned between 0.8 and 1.1 V.](/image/article/2011/PY/c0py00308e/c0py00308e-f4.gif) | ||
| Fig. 4 Typical galvanostatic charge–discharge curve of poly-3 scanned at 0.01 mA in propylene carbonate with 0.1 M [n-Bu4N]PF6 supporting electrolyte. Scanned between 0.8 and 1.1 V. | ||
Backbone oxidation
A dramatic decrease in polymer electroactivity was observed after the first cycle of the galvanostatic charge–discharge experiment, with continued degradation observed for each sequential scan (Fig. 4). This pronounced degradation and the low specific charge capacity values obtained for poly-3, led to investigations into the possible causes of degradation. A potential explanation lies in the possibility of breaks in the conjugation along the polymer chain. This may be the result of mechanical strain provoked by the ions during insertion and deinsertion21 or chemical attack by the solvent, doping ions or water contained in the electrolyte.22 Several investigations have also been carried out into the reactivity of singlet oxygen generated during UV irradiation causing degradation of PT backbones via chain breaking and disruption to the conjugation.23 It is proposed that TEMPO, a known oxidizing agent for the oxidation of alcohols, may be contributing to the accelerated oxidation of the PT backbone.Recovery of the polymer from the solutions of poly-3 and poly-1 used for electrochemistry allowed study by both ATR-IR and UV-vis spectroscopies, which offered some insight into the nature of the film degradation. As seen in the ATR-IR spectra (Fig. 5), there is a distinct new band at 1725 cm−1 in the spectrum of recovered poly-3 after electrodeposition that is not present prior to electrodeposition. In addition, it is clear from Fig. 5b, where the recovered sample of poly-3 is compared to recovered poly-1, that poly-1 does not experience the same degradation as poly-3 as no band at 1725 cm−1 is observed in this case.
 | ||
| Fig. 5 ATR-IR spectra of (a) poly-3 before (blue) and after (black) isolation from the CV solution and (b) poly-3 (black) and poly-1 (red) isolated from the polymerization reaction. | ||
The band at 1725 cm−1 is characteristic of a carbonyl stretch (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O 1700–1800 cm−1) and strongly suggests oxidation of the polymer backbone is occurring. There are several possible mechanisms that could lead to the formation of a carbonyl group within a PT backbone.23a,24 The polymer backbone may be susceptible to hydrogen abstraction from the TEMPO, and in the presence of oxygen the polymer backbone is oxidized. Scheme 2 outlines a potential mechanism for the formation of carbonyl groups leading to chain scission and reduced conjugation. UV-vis spectroscopy of recovered poly-3 also shows a blue shift in the absorption maximum (from 436 to 430 nm), directly related to a reduction in π-conjugation along the polymer backbone.
O 1700–1800 cm−1) and strongly suggests oxidation of the polymer backbone is occurring. There are several possible mechanisms that could lead to the formation of a carbonyl group within a PT backbone.23a,24 The polymer backbone may be susceptible to hydrogen abstraction from the TEMPO, and in the presence of oxygen the polymer backbone is oxidized. Scheme 2 outlines a potential mechanism for the formation of carbonyl groups leading to chain scission and reduced conjugation. UV-vis spectroscopy of recovered poly-3 also shows a blue shift in the absorption maximum (from 436 to 430 nm), directly related to a reduction in π-conjugation along the polymer backbone.
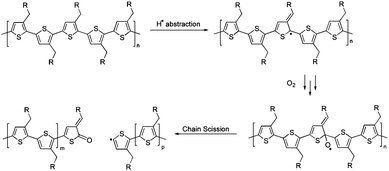 | ||
| Scheme 2 Proposed mechanism of chain scission. | ||
In the case of polymer cleavage, the TEMPO groups should still, in principle, be able to exhibit some charging behaviour. It is possible that as the conductivity of the polymer is reduced, the TEMPO groups farthest from the electrode are rendered inaccessible due to a lack of conjugation throughout the polymer. This compounds the effect of the disrupted conjugation, contributing to the lower specific capacity of poly-3.
Conclusions
A polythiophene prepared viaGRIM polymerization was functionalized with the stable free radical TEMPO. It was hoped that this new material would display improved charging and discharging capabilities for applications in organic-based batteries. The performance of the thin films in specific capacity and charge/discharge experiments was determined to be less than optimal. Further investigations suggest that the TEMPO group may be contributing to oxidation of the backbone of poly-3. The appearance of a new band in the ATR-IR spectrum at 1725 cm−1 indicates the formation of a carbonyl group, a characteristic feature of oxidized PT.24 The disruption in conjugation caused by chain scission effectively decreases the polymers ability to conduct and store charge corroborating the low capacitance.It was demonstrated that the preformed poly-3 and poly-1 were deposited on electrode surfaces such as carbon during electrochemical cycling; this is a new method of depositing soluble CPs onto active electrodes. Deposition of polymers having known structures (i.e., head-to-tail regioregularity) onto electrode surfaces provides a direct correlation between structure–property relationships observed for the surface bound species. This understanding may help improve synthetic strategies towards the desired electrochemical properties of such conjugated organic materials.
Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada for funding.References
- (a) T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS; (b) T. Suga, H. Konishi and H. Nishide, Chem. Commun., 2007, 1730–1732 RSC.
- (a) S. Patra and N. Munichandraiah, J. Appl. Polym. Sci., 2007, 106, 1160–1171 CrossRef CAS; (b) F. Marchioni, J. Yang, W. Walker and F. Wudl, J. Phys. Chem. B, 2006, 110, 22202–22206 CrossRef CAS; (c) A. Laforgue, P. Simon, C. Sarrazin and J. F. Fauvarque, J. Power Sources, 1999, 80, 142–148 CrossRef CAS; (d) A. M. P. Hussain and A. Kumar, J. Power Sources, 2006, 161, 1486–1492 CrossRef CAS; (e) A. Laforgue, P. Simon and J. F. Fauvarque, Synth. Met., 2001, 123, 311–319 CrossRef CAS.
- (a) D. M. DeLongchamp, B. M. Vogel, Y. Jung, M. C. Gurau, C. A. Richter, O. A. Kirillov, J. Obrzut, D. A. Fischer, S. Sambasivan, L. J. Richter and E. K. Lin, Chem. Mater., 2005, 17, 5610–5612 CrossRef CAS; (b) H. N. Cui, V. Teixeira, J. Zhang and H. Lee, Thin Solid Films, 2006, 515, 301–306 CrossRef CAS.
- (a) T. Aernouts, T. Aleksandrov, C. Girotto, J. Genoe and J. Poortmans, Appl. Phys. Lett., 2008, 92, 33306 CrossRef; (b) S. F. Jahn, L. Engisch, R. R. Baumann, S. Ebert and W. A. Goedel, Langmuir, 2009, 25, 606–610 CrossRef CAS; (c) V. Wood, M. J. Panzer, J. Chen, M. S. Bradley, J. E. Halpert, M. G. Bawendi and V. Bulović, Adv. Mater., 2009, 21, 2151–2155 CrossRef CAS.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–281 CrossRef CAS.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS.
- (a) A. Ma and J. M. Bobbitt, J. Org. Chem., 1991, 56, 6110–6114 CrossRef CAS; (b) A. Gheorghe, A. Matsuno and O. Reiser, Adv. Synth. Catal., 2006, 348, 1016–1020 CrossRef CAS; (c) J. Luo, C. Pardin, W. D. Lubell and X. X. Zhu, Chem. Commun., 2007, 2136–2138 RSC; (d) Z. Wang, R. Liu, Y. Jin and X. Liang, Chem.–Eur. J., 2008, 14, 2679–2685 CrossRef CAS.
- (a) M. C. Krishna, W. DeGraff, O. H. Hankovszky, C. P. Sár, T. Kálai, J. Jekö, A. Russo, J. B. Mitchell and K. Hideg, J. Med. Chem., 1998, 41, 3477–3492 CrossRef CAS; (b) M. C. Krishna, D. A. Grahame, A. Samuni, J. B. Mitchell and A. Russo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5537–5541 CAS.
- (a) L. Bucsiova, M. Yin, S. Chmela and W. D. Habicher, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 761–768 Search PubMed; (b) X. Chen, B. Gholamkhass, X. Han, G. Vamvounis and S. Holdcroft, Macromol. Rapid Commun., 2007, 28, 1792–1797 CrossRef CAS.
- (a) Y. Takahashi, N. Hayashi, K. Oyaizu, K. Honda and H. Nishide, Polym. J. (Tokyo, Jpn.), 2008, 40, 763–767 CrossRef CAS; (b) K. Oyaizu, T. Suga, K. Yoshimura and H. Nishide, Macromolecules, 2008, 41, 6646–6652 CrossRef CAS; (c) H. Nishide, S. Iwasa, Y.-J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- J. Finden, T. K. Kunz, N. R. Branda and M. O. Wolf, Adv. Mater., 2008, 20, 1998–2002 CrossRef CAS.
- R. S. Loewe, S. M. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250 CrossRef CAS.
- H. G. Aurich, in Nitrones, Nitronates and Nitroxides, ed. S. Patai and Z. Rappoport, John Wiley & Sons, Chichester, 1989, vol. 2, pp. 313–370 Search PubMed.
- A. Gheorghe, E. Cuevas-Yañez, J. Horn, W. Bannwarth, B. Narsaiah and O. Reiser, Synlett, 2006, 2767–2770 CAS.
- C. Morat and A. Rassat, Tetrahedron, 1972, 28, 735–740 CrossRef CAS.
- (a) K. S. Jang, H. C. Ko, B. Moon and H. Lee, Synth. Met., 2005, 150, 127–131 CrossRef CAS; (b) M. Laferrière, R. E. Galian, V. Maurel and J. C. Scaiano, Chem. Commun., 2006, 257–259 RSC.
- C. N. Banwell and E. M. McCash, in Fundamentals of Molecular Spectroscopy, McGraw-Hill Publishing Company, Berkshire, 4th edn, 1994, pp. 245–256 Search PubMed.
- C. C. Whisnant, S. Ferguson and D. B. Chesnut, J. Phys. Chem., 1974, 78, 1410–1415 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Hoboken, NJ, 2001 Search PubMed.
- A. Laforgue, P. Simon, C. Sarrazin and J.-F. Fauvarque, J. Power Sources, 1999, 80, 142–148 CrossRef CAS.
- J. Wang, Electrochim. Acta, 1994, 39, 417–429 CrossRef CAS.
- J. Wang, Electrochim. Acta, 1997, 42, 2545–2554 CrossRef CAS.
- (a) M. Manceau, A. Rivaton, J.-L. Gardette, S. Guillerez and N. Lemaître, Polym. Degrad. Stab., 2009, 94, 898–907 CrossRef CAS; (b) M. Manceau, A. Rivaton and J.-L. Gardette, Macromol. Rapid Commun., 2008, 29, 1823–1827 CrossRef CAS.
- (a) S. Holdcroft, Macromolecules, 1991, 24, 4834–4838 CrossRef; (b) M. S. A. Abdou and S. Holdcroft, Macromolecules, 1993, 26, 2954–2962 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR and ESR spectra, cyclic voltammetry of poly-3. See DOI: 10.1039/c0py00308e |
| This journal is © The Royal Society of Chemistry 2011 |