Synthesis of polyphosphorinanes Part II.† Preparation, characterization and thermal properties of novel flame retardants
Claire
Negrell-Guirao
a,
Bernard
Boutevin
*a,
Ghislain
David
a,
Alain
Fruchier
a,
Rodolphe
Sonnier
b and
José-Marie
Lopez-Cuesta
b
aInstitut Charles Gerhardt UMR 5253, IAM, Rue de l'école normale, 34296, MONTPELLIER cedex 5, France. E-mail: bernard.boutevin@enscm.fr; Fax: +0467147220; Tel: +0467144307
bCentre des Matériaux de Grande Diffusion. Ecole des Mines d'Alès, 6 av. Clavières, 30319, ALES, France. E-mail: jose-Marie.lopez-Cuesta@ema.fr; Fax: +0466785365; Tel: +0466785334
First published on 14th October 2010
Abstract
We report here the investigation of new flame retardant compounds for textiles, obtained by radical polymerization of allyloxydioxaphosphorinanes. These monomers were first synthesized by a transesterification reaction. A deep NMR study of such compounds revealed the existence of diastereoisomers Z and E. Radical polymerizations of allyloxydioxaphosphorinanes in the presence of a chain transfer agent, i.e.dimethylhydrogenophosphonate, led to oligomers of rather low molecular weights and especially adducts with DP 1 and 2. It was demonstrated that radical polymerization of allyloxydioxaphosphorinanes with P–R (R being alkyl or aryl) does afford only linear oligomers instead of hyperbranched polymers when R is hydrogen. The thermogravimetric analyses of the oligomers showed a good thermal stability and a high content of char residue (more than 16 wt%). The flame retardant properties were also evaluated by microcalorimeter tests and revealed an efficient behavior especially in the condensed phase, comparatively to Antiblaze 19®, the flame retardant commercial product for polyester textile.
Introduction
Since a few years ago, flame retardants (FRs) for textiles have regained much interest since the introduction of new European legislation, especially regarding REACH. Indeed, halogenated FRs are now recognized as highly toxic compounds and have been replaced by non-toxic FRs such as phosphorus-based FRs.1–3 These phosphorus-based flame retardants additives are introduced into materials by two methods: on one hand either as additives in melted polymers or applied as end treatment, on the other hand as reacting agents during the polymer synthesis. In the particular frame of textiles with poly(ethylene terephtalate) (PET), Antiblaze 19® (marketed by the Mobil Company) is used as a FR.4 This very effective compound is a mixture of two cyclic phosphite structures. This industrial product is the reference in the fireproofing of the PET; its effectiveness as a flame retardant is ascribed to the phosphoric esters which entail transesterification reactions with PET. The aim of this work is to synthesize an equivalent macromolecular product of Antiblaze, starting from a similar cyclic structure which is compatible with PET. In the first article,5 the synthesis of polymers obtained from 5-ethyl-5-(allyloxymethyl) 2-oxo-1,3,2-dioxaphosphorinane was described (Scheme 1-A). Its radical polymerization leads to two different species according to the polymerization type: polyaddition of the monomer (Scheme 1-B) and linear polymerization of the double allylic bond (Scheme 1-C). Both mechanisms take place simultaneously leading to hyperbranched structures, which have poor solubility in conventional organic solvents. Telomerization of 5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane was also suggested to decrease the branching rate. The monoadduct, i.e. degree of polymerization (DP) of 1, or the telomer, i.e. DP higher than 1, could be obtained and were used as effective products in fireproofing of PET textiles. However their low molecular weights generate migration towards the surface; this additive-exudation creates a change in the mechanical properties and/or anti-fire properties and even a risk of pollution. FR development in polymer form should allow reduction of this phenomenon. To our knowledge, only Friedman6 polymerized dioxaphosphorinane bearing allyl ether groups by using radical initiators such as azoic or peroxides types, however neither molecular weight values nor yields of the synthesized polymers were reported. | ||
| Scheme 1 Polymerization of 5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane by radical solution. | ||
In this paper, allyl ether monomers as well as the corresponding monoadducts have been synthesized in which the reactive P–H bond was replaced by either alkyl or aryl group fixed to the phosphorus atom to avoid the formation of hyperbranched structures. The radical polymerizations of these monomers have been carried out in the presence of a chain transfer agent and the fire retardant properties have been evaluated as a function of their molecular weights.
Experimental
Materials
All the solvents and reagents (Sigma-Aldrich, Fluka or Acros) were used with a purity of 98–99%. 1,3-Propanediol, diethyl hydrogenophosphonate, dimethyl hydrogenophosphonate, allyl butyl ether, 2-(allyloxymethyl)-2-ethyl-1,3-propanediol, allyl bromide, sodium hydride (60% in oil), triethyl phosphite, tert-butyl potassium oxide, 2,5-bis(tert-butylperoxy)-2,5-dimethylhexane (VAROX) and tert-amyl peroxypivalate (TAPPI) were used without purification. 2,2′-Azobis(isobutyronitrile) (AIBN) was purified by recrystallization from methanol and dried under vacuum.1H and 31P NMR spectra were recorded in CDCl3 or acetone d6 solutions (between 5–10 mg of each compound dissolved in 0.6 ml of solvent) on a Bruker Avance 400 MHz spectrometer. 1H and 31P chemical shifts are expressed in ppm/TMS and ppm/H3PO4 external standard, respectively. Standard Bruker gradient versions of 2D correlation sequences were used: NOESYPH, HMQC 1H-13C. Solutions for NOESY experiments were degassed with argon. All non-first-order spectra were calculated with g-NMR. Size exclusion chromatography (SEC) was performed on a Varian apparatus equipped with a RI Shodex refractive index detector. Two PL-gel mix D columns were used at 70 °C with a 0.8 ml min−1 flow rate of DMF, calibrated using polymethylmethacrylate standards. TGA experiments were conducted on a Pyris 1 TGA by Perkin-Elmer. They were performed on 2–10 mg samples under nitrogen at a heating rate of 10 °C min−1. Thermogravimetric analysis was coupled to Fourier transform infrared spectroscopy TGA/IRTF (IFS660 Bruker). The microscale combustion calorimeter (MCC) tests were carried out on a Fire Testing Technology apparatus, which is a pyrolysis-combustion flow calorimeter. 2–3 mg samples were heated to 750 °C at a heating rate 1 K s−1 in an inert gas steam. Then the pyrolysis products were mixed with oxygen prior to entering a 900 °C combustion furnace and the heat of combustion were measured by oxygen consumption principle.
Monomers and polymers syntheses
1H NMR (400 MHz, CDCl3, δ): 0.85 (t, H2′′(B)); 0.90 (t, H2′′(A)); 1.20 (t, H2′′′(B)); 1.21 (t, H2′′′(A)); 1.39 (q, H1′′(B)); 1.56 (q, H1′′(A)); 1.84 (q, H1′′′(B)); 1.85 (q, H1′′′(A)); 3.36 (s, H1′(A)); 3.49 (s, H1′(B)); 3.95 (d, H3′(A)); 4.00 (d, H3′(B)); 3.90–4.41 (m, H4(A + B)-H6(A + B)); 5.18–5.27 (m, H5′(A + B)); 5.85 (m, H4′(A)); 5.88 (m, H4′(B)).
1H NMR (400 MHz, CDCl3, δ): 0.84 (t, H2′′(B)); 0.89 (t, H2′′(A)); 0.91 (t, H4′′′(A + B)); 1.25–1.80 (m, H1′′′ 2′′′ 3′′′(A + B)); 1.39 (q, H1′′(B)); 1.54 (q, H1′′(A)); 3.36 (s, H1′(A)); 3.48 (s, H1′(B)); 3.94 (d, H3′(A)); 4.00 (d, H3′(B)); 3.90–4.39 (m, H4(A + B)-H6(A + B)); 5.16–5.27 (m, H5′(A + B)); 5.84 (m, H4′(A)); 5.88 (m, H4′(B))
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethyl acetate/heptane and gradually increasing to 1:0 ethyl acetate/heptane to give the monomer as a colorless oil (Yield: 57%).
:1 ethyl acetate/heptane and gradually increasing to 1:0 ethyl acetate/heptane to give the monomer as a colorless oil (Yield: 57%).
1H NMR (400 MHz, CDCl3, δ): 0.66 (t, H2′′(A)); 0.76 (t, H2′′(B)); 1.12 (q, H1′′(A)); 1.28 (q, H1′′(B)); 2.94 (s, H1′(B)); 3.23 (t, H1′′′(B)); 3.26 (t, H1′′′(A)); 3.30 (s, H1′(A)); 3.65 (d, H3′(B)); 3.89 (d, H3′(A)); 3.75–4.30 (m, H4(A + B)-H6(A + B)); 5.11–5.19 (m, H5′(A + B)); 5.75 (m, H4′(A)); 5.79 (m, H4′(B)); 7.10–7.28 (m, Haromatics(A + B))
| Run | Monomer | C0 (initiator)a | T/°C | R0b | S0c | αMd | DPne |
|---|---|---|---|---|---|---|---|
| a C0 = [initiator]0/[monomer]0. b R0 = [CTA]0/[monomer]0. c S0 = [solvent]0/[monomer]0. d conversion obtained from 1H NMR. e obtained by SEC. | |||||||
| 1 | B | 0.050 (VAROX) | 115 | 0 | 0 | 0.17 | — |
| 2 | B | 0.100 (VAROX) | 110 | 0.52 | 0 | 0.18 (48 h) | 2.2 |
| 3 | C | 0.053 (AIBN) | 70 | 0.50 | 3.8 | 0.11 (24 h) | 2.3 |
| 4 | C | 0.050 (AIBN) | 70 | 0.96 | 3.9 | 0.18 (24 h) | 2 |
| 5 | C | 0.050 (AIBN) | 70 | 2.10 | 3.9 | 0.57 (24 h) | 1.6 |
| 6 | C | 0.050 (VAROX) | 110 | 8 | 0 | 0.90 (2 h) | 1.1 |
Results and discussion
Synthesis of dioxaphosphorinane monomers
As mentioned in the Introduction, radical polymerization of 5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane leads to a mixture (Scheme 1) of both oligomers (Mn of about 800 g mol−1) and hyperbranched high molecular weight polymers (Mn of about 100,000 g mol−1), ascribed to the high reactivity of the P–H bond from the monomer.To avoid this behavior, the radical polymerization of dioxaphosphorinanes bearing P-alkyl or P-aryl groups has been carried out. First of all, the synthesis of these monomers was investigated. Several techniques, described in the literature, allow the synthesis of a dioxaphosphorinane monomer with an alkyl or aryl group attached to the phosphorus atom. Usually, dioxaphosphorinane monomers can be obtained by direct transesterification of a diol with an alkylphosphonate. Transesterification of a diol, i.e.2-allyloxymethyl-2-ethyl-1,3-propanediol, with diethyl ethylphosphonate (commercial product) was first carried out at 140 °C (Scheme 2A), however even after 8 h no ethanol elimination was observed.
 | ||
| Scheme 2 Syntheses of dioxaphosphorinanes bearing P-alkyl or P-aryl groups | ||
Indeed, this reaction usually requires a tautomeric equilibrium between PIII-PV since the substitution is enhanced in the PIII form. This tautomeric equilibrium only exists with a P–H bond (Scheme 3).7,8
 | ||
| Scheme 3 Tautomeric equilibrium of phosphonate | ||
In the case of phosphonates with P–R (R ≠ H), basic catalysis is required.9 The use of tBuOK in stoichiometric proportions, according to two others reagents, activates the alcohol group and allows to form 2,5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane (Scheme 2A). The colorless oil was obtained in 20% yield after purification, with a very good purity as demonstrated by 1H NMR (Fig. 1A). The disappearance of the hydrogen linked to the phosphorus atom centered at 6.89 ppm and the appearance of the characteristic peaks of the ethyl group (1.17 ppm for CH3 and 1.82 ppm for the CH2 with coupling constants from 18 to 20 Hz) were observed. The low yield and the low choice of commercial dialkyl-alkylphosphonate however confirm the lack of interest of this procedure.
 | ||
| Fig. 1 1H NMR spectra of 2,5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane (below), 2-butyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane (centre) and 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane (above). | ||
To obtain the corresponding dioxaphosphorinane with R = butyl or benzyl, another procedure was used (Scheme 2B). An effective way uses caesium carbonate and tetrabutylammonium iodide10 to promote the phosphonate anion generated in situ from dialkyl hydrogenophosphonate followed by its alkylation with a alkyl halide. 11,12 The synthesis of dioxaphosphorinane with R = butyl or benzyl then requires two steps, the first one is the synthesis of the already described 5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane5 followed by the modification of the P–H bond in the presence of a weak base (carbonate type) Cs2CO3 (Scheme 2B). After 24–72 h of reaction and a flash chromatography purification, phosphonates (with R = benzyl or butyl) were obtained in approximately 60% yield. Their structures and their purity were confirmed by 1H NMR (Fig. 1B and C) by the disappearance of the characteristic peak assigned to the proton linked to the phosphorus atom centered at 6.89 ppm (1J (P–H) = 673.8 and 683.6 Hz for 2 diastereoisomers) and also by the occurrence of the characteristic peaks for butyl (B) (the CH3 in 0.91 ppm and the CH2 between 1.25–1.80 ppm) or benzyl (C) (7.10–7.26 ppm for the aromatic protons). In 31P NMR, a splitting of diastereoisomer peaks (4.04 and 4.50 ppm for the dioxaphosphorinane P–H) to 29.73 and 31.34 ppm for the butyl group and to 21.81 and 24.37 ppm for the benzyl group linked to the phosphorus atom was observed.
As mentioned above, these rather complex NMR data demonstrate the existence of two diastereoisomers Z and E due to the two pseudo-asymmetric centers for each monomer; these two diastereoisomers were not separated during the polymerization process. General 31P-NMR properties of 2-oxo-1,3,2-dioxaphosphorinanes have been reviewed by Maryanoff et al.13 and Gorenstein.14 The influence of their conformation on 1H and 31P spectra was more specifically reviewed by Bentrude and Setzer.15–17
In order to assess the proportions of each diastereoisomers NOESY experiments were conducted. As an example, NOESY spectra of isomers from 2,5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane are given in Fig. 2 and their conformations are shown in Scheme 4.
 | ||
| Fig. 2 Partial NOESY spectra of the two isomers of 2,5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane in CDCl3. | ||
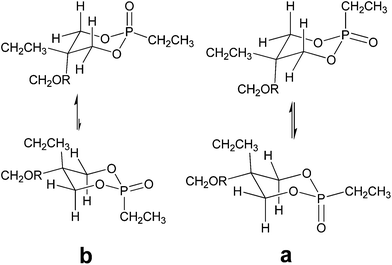 | ||
| Scheme 4 Structures of the two isomers a and b of 2,5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane | ||
Concerning the stereoisomer b, the OCH2 on C5 (H1′′ from Fig. 1) is only correlated with the equatorial H4/H6 while the CH2CH3 (H1′) is correlated with both axial and equatorial H4/H6. This is in favour of an axial preference for the OCH2 group. For isomer a results are less clear since both methylene groups on C5 are correlated with the four protons on C4/C6. Moreover a dipolar interaction is observed between the PCH2CH3 (H1′′′) and the H4/H6protons at 3.91 ppm (Fig. 2, a1). This interaction is absent in compound b (Fig. 2, b1). These observations suggested the structures and equilibria depicted in Scheme 4. These results show that two chair forms exist in rapid equilibrium. For isomers b, one chair form is predominant while for isomers a, the equilibrium takes place between conformers almost equally populated. In all cases the observed 3JPOCH are weighted by means of the limiting values. The hypothesis that chair forms are involved and not boat, twisted or any other mobile structures is strengthened by the fact that a 4JHH is always observed between H4 and H6.
Moreover, the appearance of the AA′MM′X spin system is found to be dependent on the populations of each conformer at equilibrium. The external transitions of the pseudo-triplets are sensitive to the existence of more than one chair form.
In the conditions of rapid equilibrium between I and II, the observed coupling constants are given by (Fig. 3):
| JAA′ = p × J1 + (1 − p)×J′2 |
| JMM′ = p × J2 + (1 − p)×J′1 |
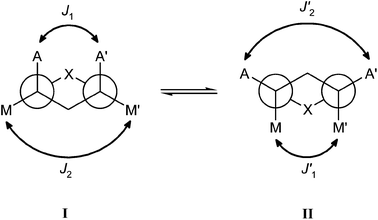 | ||
| Fig. 3 Newman representation of the equilibrium between the two limiting chair forms I (population: p) and II (population: 1 − p) of dioxaphosphophorinane compound. | ||
If we assume that J1 ≅ J′1 and that J2 ≅ J′2, the coupling constants of the two AB subspectra are:
| K = JMM′ + JAA′ = J2 + J1 |
| M = JMM′ − JAA′ = (J2 − J1) × (2 × p − 1) |
The subspectrum K is not independent of p. If the 4J between axial protons (J1) is close to zero, the values of M are obvious in two cases.
The first one was already encountered with compound 1 in which p ≅ 1. In this case, K = M = J2, the two subspectra K and M are superposed and the pseudo triplets show intensities close to 1:2:1.
In the second limiting case, none of the two chairs are prevalent and p ≅ 0.5 with the consequence that M ≅ 0. The subspectrum M becomes a AX subspectrum with JAX = 0 and its two lines are superposed to the large transitions of the two A2 subspectra separated by L = N = JAM + JAM′ with JAM = Jgem and JAM′ ≅ 0. The intensities of the pseudo triplets are in this case 0.5:3:0.5. This is illustrated in Fig. 4 where the experimental spectrum a of a mixture of the two isomers Z and E of butyl compound is compared to two simulated spectra b and c in which p = 1 and 0.5 respectively. Obviously, the experimental spectrum of 3a (signals at 4.39 and 3.90 ppm) is closer to spectrum c than to spectrum b.
 | ||
| Fig. 4 Experimental (a) and calculated spectra (b, p = 1; c, p = 0.5) of the two isomers of compound 3. | ||
The evolution of 3JPOCH when the temperature is decreased is an indication of the equilibrium displacement which should have an effect on the K and M subspectra. Unfortunately, the resolution at low temperature was not good enough to allow a discussion of the shape of the pseudo-triplet external transitions.
Radical polymerization of dioxaphosphorinane monomers
The conventional radical polymerization of dioxaphosphorinane monomers was described in a 1960s patent,6 however, very low molecular weights could be only achieved, as for us. In the first paper the radical polymerization of 5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane, i.e. with P–H bond, led to high molecular weight polymers with high yields, but hyperbranched polymers were obtained.5 First of all, homopolymerization of dioxaphosphorinane monomers (B) and (C) was then carried out without the use of a chain transfer agent (Table 1), however only poor yields could be achieved, probably due to the steric hindrance of the dioxaphosphorinane monomers. Under the free-radical conditions, the homopolymerization of allyl monomers is very unlikely, and when reaction occurs, it usually polymerizes at rather low rates.18 This behavior is the result of a degradative chain transfer; the weakness of the allyl C–H bond arises from the high resonance stability of the allylic radical, which is too stable to reinitiate polymerization and undergoes termination by reaction with propagating radicals.19Radical polymerizations of dioxaphosphorinane monomers were then carried out in the presence of dimethylhydrogenophosphonate HP(O)(OMe)2 as chain transfer agent (CTA) (Scheme 5). | ||
| Scheme 5 Radical polymerizations of dioxaphosphorinane monomers in the presence of HP(O)(OMe)2 as CTA. | ||
As previously demonstrated,5 this CTA is reactive towards the allyl double bond and also allows incorporation at a phosphonate functionality one chain-end, which will enhance the expected fire retardant properties. Radical polymerizations were carried out in different reaction conditions, gathered in Table 1. Table 1 clearly shows that the monomer conversion is mainly influenced by the concentration of CTA, i.e. R0, whatever the reaction conditions. Monomer conversions of 60% could be reached (run 4) only for R0 higher than 2. This behavior is the result of an apparent high CT (chain transfer constant) value due to the low propagation constant value of allyl monomers. This is also proved by the low degree of polymerizations DPn obtained by SEC. The SEC distributions of crude products obtained from runs 3 to 6 (Table 1) are displayed in Fig. 5 for 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane telomers, i.e. oligomers obtained with a CTA.
 | ||
| Fig. 5 SEC distributions of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane oligomers (crude) obtained in the presence of a CTA. | ||
From the SEC distributions the first peak was assigned to the monoadduct (DPn equal to 1), and the second one assigned to the diadduct (DPn equal to 2). The ratio of monoadduct to diadduct is dependant on R0. For R0 of 8 (run 6), mainly the monoadduct was obtained. This result proves that the replacement of H of P–H bond by alkyl or aryl group leads to linear oligomers, however only very low DPn can be reached.
Flame retardant properties of dioxaphosphorinane oligomers
Phosphorus containing FR are known to be able to act both in the vapor phase and in the condensed phase.20,21 In the vapor phase volatile radicals, such as PO2·, PO· or HPO·, act as oxygen inhibitors.22 In the condensed phase, they may create, after degradation, a char residue acting as barrier for the polymer matrix. First of all, thermogravimetric analyses (TGA) were performed on 2–10 mg samples under nitrogen atmosphere at a heating rate of 10 °C min−1, from 50 to 600 °C. Fig. 6 shows the TGA results of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane (C) and the corresponding monoadduct and diadduct obtained by radical polymerization in the presence of CTA (runs 3 and 6 from Table 1), the thermogram of Antiblaze 19® is also given. Thermograms of dioxaphosphorinane oligomers bearing alkyl groups are not shown here since their degradation is lower than that of the aryl homologues, as already mentioned by Hoang et al.23,24 on bicyclic phosphonate FR. First, compared to its adduct obtained with dimethyl hydrogenophosphonate CTA the monomer shows a depletion both on the thermal stability and on the content of residue and a similar trend than that of Antiblaze 19®. This result enhances the use of the CTA especially to provide a higher content of phosphorus atom for the FR content and higher molecular weight. | ||
| Fig. 6 Thermograms (recorded under N2) of both monoadduct and diadduct obtained by radical polymerization of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane in the presence of CTA (runs 3 and 6) and Antiblaze 19®. | ||
The Tonset, starting degradation temperature, as well as T10%, 10% decomposition temperature, are very similar for both the mono and diadduct; Tonset is showed at 230 and 240 °C for the mono and diadduct, respectively, whereas T10% is measured at 270 and 265 °C for the mono and diadduct, respectively. The weight fractions of charred residue are different for both compounds since about 16 wt% and 6 wt% are obtained for the mono and diadduct, respectively. If these FRs are incorporated in a no-charring polymer matrix, the vapor phase action can be considered as the primary mechanism of the action; but they leave about 20% of charred residue above 600 °C, indicating that condensed phase mechanism may also contribute. The weight fraction of residue is not directly connected to the amount of phosphorus as the diadduct shows a slightly higher amount of phosphorus than the corresponding monoadduct. Compared with those of others dioxaphosphorinane products from the literature,23 similar char yields were observed. The nature of this char was composed of phosphonated group associations like pyrophosphonic acid.25 Interestingly, the TGA traces show multi-stage decompositions for both compounds. The first decomposition is explained by the break of P–O–C26 bonds of methyl phosphonic chain, then above 300 °C, there is a loss of the carbon chain, leading to the formation of pyrophosphoric acid and finally above 375 °C the loss of benzyl group. A TGA/FTIR experiment was carried out to analyze the volatile products obtained during the degradation at three different temperatures, i.e. 285, 356 and 371 °C (Fig. 7). In the first stage of the monoadduct degradation, i.e. 285 °C, the loss of methanol is observed (νOH 3250 cm−1, νCsp3H 2975 cm−1 and νC–OH 1064 cm−1). At 356 °C a loss of aldehyde groups is observed (νCsp3H 2975 cm−1, and νC = O 1718 cm−1) followed by the loss of carbonated molecules (νCsp3H 2975 cm−1, νCH2 1461 cm−1). Then at 371 °C the νCsp2–H vibration is visible around 3100 cm−1 which demonstrates the loss of aromatic group.
 | ||
| Fig. 7 TGA/FTIR of monoadduct obtained by radical polymerization of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane in the presence of CTA (run 6). | ||
Finally, microcalorimeter tests have been performed on both adducts of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane, showing the evolution of the heat release rate (HRR) with the temperature (Fig. 8). The sum heat release capacity (HRC) and the total heat release (THR) are more representative of the FR capacity; these parameters can be reached from the HRR evolution vs. temperature (Fig. 8), i.e. sumHRC is obtained from sum of the deconvoluted peaks of HRR divided by the heat rate of the sample, whereas THR represents the total area of the HRR. The lower the THR and sumHRC, the better the FR properties.
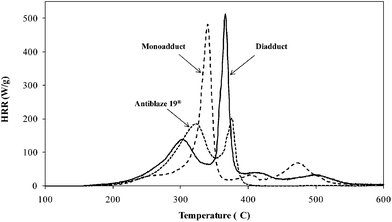 | ||
| Fig. 8 Heat release rate (HRR) vs. temperature for both monoadduct and diadduct obtained by radical polymerization of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane in the presence of CTA (runs 3 and 6) and for Antiblaze 19®. | ||
The monoadduct of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane shows one main peak whereas two main degradation can be observed from diadduct combustion. Further, a small degradation is also observed above 400 °C corresponding to the decomposition of the benzyl group. The evolution of HRRvs. temperature is also given for Antiblaze 19® and also two main degradations are obtained resulting from both structures, i.e. mono and diadduct (Scheme 6).
 | ||
| Scheme 6 Structures of Antiblaze 19® | ||
sumHRC and THR values are given in Table 2 for the monomer and the corresponding adducts and compared to those obtained for Antiblaze 19®, marketed as FRs.4 It is seen that both the THR and sumHRC values are higher than that of Antiblaze 19®. The same remark can be drawn from the peak temperature, the higher the peak temperature the better the FR behavior. According to these results adducts of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane as well as the monomer itself show a lower behavior of FR than that of Antiblaze 19®; this might be probably related to the content of phosphorus atom (expressed in Table 2), which enhances the resistance to combustion with Antiblaze 19®. However the low Mw of Antiblaze 19® remains a drawback and the higher Mw for adducts will probably allow avoiding of migration into the polyester textile. These adducts seem also to behave differently in the condensed phase. Indeed, whereas Antiblaze 19® shows only a poor residue char rate, adducts of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane give until 16% of char residue, which demonstrates the potential action of the compounds in the condensed phase.
| Compound | Mw/g mol−1 | T a/°C | THR b/kJ g−1 | sumHRCc | % P (wt%) | Residued (%) |
|---|---|---|---|---|---|---|
| a Peak temperature. b Total heat release. c Heat release capacity. d Residue from TGA. | ||||||
| Monomer | 310 | 315 | 24.9 | 408 | 10.3 | 5.8 |
| Monoadduct | 450 | 340 | 18.3 | 578 | 15.0 | 16.0 |
| Diadduct | 600 | 366 | 22.0 | 686 | 16.0 | 6.0 |
| Antiblaze 19® | 331 | 375 | 16 | 351 | 22.0 | 1.5 |
Conclusions
New allyloxydioxaphosphorinanes have been efficiently synthesized; these compounds being phosphonate type monomers with P–R. It was demonstrated that when R is not hydrogen, i.e. alkyl or aryl, hyperbranched polymers can be avoided by radical polymerization. In the presence of a CTA also bearing a phosphonate group, adducts of dioxaphosphorinanes were obtained with good yields. Compared to the commercial flame retardant compound used for textiles, i.e. Antiblaze 19®, the adducts of dioxaphosphorinane, and especially of 2-benzyl-5-ethyl-5-(allyloxymethyl)-2-oxo-1,3,2-dioxaphosphorinane, show a better behavior in the condensed phase since more than 16% char residue were obtained at 600 °C, whereas Antiblaze 19® only gives less than 2% of char residue. Further, the molecular weights of the synthesized adducts are higher than that of Antiblaze 19®, which will probably avoid migration of the FR towards the surface of the textile. However, it was shown that in the gas phase, FR properties of adducts are lower than the commercial product, for instance both THR and HRC values were found to be higher than that of Antiblaze 19®. This may be assigned to the chemical structure of Antiblaze 19®, which is a phosphate-type FR. This assumption could be checked by synthesizing new allyloxydioxaphosphorinanes with P–O–R (R being alkyl or aryl), this will be the subject of a forthcoming work.Notes and references
- S. V. Levchik and E. D. Weil, Adv. Fire Retard. Mater., 2008, 41–66 Search PubMed.
- E. D. Weil and S. V. Levchik, J. Fire Sci., 2008, 26, 243–281 Search PubMed.
- S. V. Levchik and E. D. Weil, J. Fire Sci., 2006, 24, 345–364 Search PubMed.
- A. R. Horrocks, Rev. Prog. Color., 1986, 16, 62–101 Search PubMed.
- C. Negrell-Guirao and B. Boutevin, Macromolecules, 2009, 42, 2446–2454 CrossRef CAS.
- L. Friedman, FR Pat. 1375860, 1964.
- K. H. Worms and M. Schmidt-Dunker, Org. Phosphorus Compd., 1976, 7, 1–486 Search PubMed.
- D. E. Ailman and R. J. Magee, Org. Phosphorus Compd., 1976, 7, 487–871 Search PubMed.
- J.-N. Volle, D. Virieux, M. Starck, J. Monbrun, L. Clarion and J.-L. Pirat, Tetrahedron: Asymmetry, 2006, 17, 1402–1408 CrossRef CAS.
- R. J. Cohen, D. L. Fox, J. F. Eubank and R. N. Salvatore, Tetrahedron Lett., 2003, 44, 8617–8621 CrossRef CAS.
- A. Michaelis and T. Becker, Ber. Dtsch. Chem. Ges., 1897, 30, 1003–9 CrossRef.
- A. F. McKay, R. O. Braun and G. R. Vavasour, J. Am. Chem. Soc., 1952, 74, 5540–1 CrossRef CAS.
- B. E. Maryanoff, R. O. Hutchins and C. A. Maryanoff, Stereochemistry, 1979, 11, 187–326 Search PubMed.
- D. G. Gorenstein, Prog. Nucl. Magn. Reson. Spectrosc., 1983, 16, 1–98 CrossRef.
- W. G. Bentrude and W. N. Setzer, Methods Stereochem. Anal., 1987, 8, 365–89 Search PubMed.
- W. G. Bentrude, Conform. Behav. Six-Membered Rings, 1995, 245–93 Search PubMed.
- W. G. Bentrude, Phosphorus-31 NMR Spectral Prop. Compd. Charact. Struct. Anal., 1994, 41–53 Search PubMed.
- P. D. Bartlett and R. Altschul, J. Am. Chem. Soc., 1945, 67, 816–22 CrossRef CAS.
- I. Cho and J. Kim, Polymer, 1998, 40, 1577–1580.
- S. V. Levchik and E. D. Weil, Polym. Int., 2005, 54, 11–35 CrossRef CAS.
- F. Laoutid, L. Bonnaud, M. Alexandre, J. M. Lopez-Cuesta and P. Dubois, Mater. Sci. Eng., R, 2009, 63, 100–125 CrossRef.
- S. Duquesne, J. Lefebvre, G. Seeley, G. Camino, R. Delobel and M. Le Bras, Polym. Degrad. Stab., 2004, 85, 883–892 CrossRef CAS.
- D. Hoang and J. Kim, Polym. Degrad. Stab., 2008, 93, 36–42 CrossRef CAS.
- D. Hoang, J. Kim and B. N. Jang, Polym. Degrad. Stab., 2008, 93, 2042–2047 CrossRef CAS.
- D. D. Jiang, Q. Yao, M. A. McKinney and C. A. Wilkie, Polym. Degrad. Stab., 1999, 63, 423–434 CrossRef CAS.
- Y.-L. Liu, G.-H. Hsiue, Y.-S. Chiu, R.-J. Jeng and L.-H. Perng, J. Appl. Polym. Sci., 1996, 61, 613–621 CrossRef CAS.
Footnote |
| † For Part I see ref. 5. |
| This journal is © The Royal Society of Chemistry 2011 |