DOI:
10.1039/C0PY00282H
(Paper)
Polym. Chem., 2011,
2, 229-235
Synthesis and use of a new alkene-functionalized SG1-based alkoxyamine†
Received
31st August 2010
, Accepted 7th October 2010
First published on 29th October 2010
Abstract
A new alkene-functionalized SG1-based alkoxyamine was synthesized and used to prepare well-defined functional polymers by nitroxide mediated polymerization. The latter were characterized by NMR, SEC and MALDI-TOF mass spectrometry. Livingness was assessed by chain extension toward AB-type block copolymer. The alkene functionality located at the α-chain-end allowed the post-modification of the polymer by thiol–ene coupling reaction.
Introduction
Controlled Radical Polymerization (CRP) processes attract much attention because they provide access to polymeric materials with well-defined compositions, narrow molar mass distributions, low polydispersities and sophisticated architectures.1 Among the various processes for CRP, Nitroxide Mediated Polymerization2 (NMP) based on the use of stable nitroxide radicals (mainly 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO), N-tert-butyl-N-[1-phenyl-2-methylpropyl] nitroxide (TIPNO) and N-tert-butyl-N-[1-diethoxyphosphoryl-2,2-dimethylpropyl] nitroxide (SG1)) has emerged because it is very simple and requires, in most cases, only the use of an appropriate alkoxyamine for the polymerization reaction. The development of an SG1-based alkoxyamine with a tertiary 2-(hydroxycarbonyl)prop-2-yl initiating radical (e.g. BlocBuilder® from Arkema) allowed a broad variety of monomers to be polymerized in a controlled/living fashion.3–8 In addition, the carboxylic acid group is a convenient and versatile reactive group for the design of other types of functional alkoxyamines.9,10 For example, Vinas et al.11 described the preparation of a BlocBuilder® derivative with an N-succinimidyl ester (MAMA–NHS, Scheme 1), which was shown to be an excellent initiator for NMP and allowed the formation of well-defined polymers. This MAMA–NHS initiator can be particularly convenient for the post-functionalization of polymer chain ends. Recently, our group12 and Chevigny and co-workers13 successfully used this alkoxyamine to graft polymer chains onto primary amine-coated silica particles.
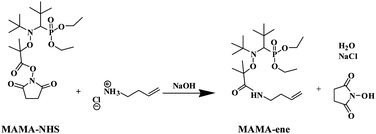 |
| | Scheme 1 Synthesis of the alkene-functionalized SG1-based alkoxyamine from the activated ester MAMA–NHS. | |
In this spirit, the aim of this work was to propose a strategy for the synthesis of a new functional alkoxyamine derived from BlocBuilder® by taking advantage of the high reactivity of MAMA–NHS and the simple conditions and high yield of the reaction with primary amine-based molecules. We chose to incorporate a non-activated alkene group by reacting MAMA–NHS with 3-butenylamine. Based on previous studies described in the literature and performed under RAFT14 and ATRP15,16 conditions, this type of double bond has sufficiently low reactivity in radical polymerization and will not be consumed. Although the synthesis of alkene-functionalized SG1-based or TEMPO-based alkoxyamine bearing a secondary alkyl radical as an initiating species has already been reported in the literature, the synthetic strategy was different and their use in polymerization experiments has not been described.9,10 Indeed, only macromonomers in which the vinyl terminal group was introduced at the end of the polymerization17 or by post-modification18 have been reported by NMP.
The idea was to elaborate well-defined, α-functional polymersviaNMP with an end-group allowing simple and efficient coupling reaction. Among the several strategies that can be envisioned, we opted for the thiol–ene coupling reaction (hydrothiolation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond) because it proceeds under mild reaction conditions and is tolerant to a large number of functional groups.19 Moreover, it is a click reaction that does not require metal catalyst in comparison to the copper-catalysed azide–alkyne click reaction which is an advantage for biological applications.20 In addition, our aim was to introduce an end-group compatible with other reactive groups incorporated along the polymer chains by copolymerization, allowing the synthesis of multifunctional polymers in a single copolymerization step. Those functional groups were chosen for their ability to react with primary-amine-functionalized substrates under mild conditions, without interference with the α-end-group. They were chloromethyl (from vinylbenzyl chloride) and N-succinimidyl ester (from N-succinimidyl acrylate).
C bond) because it proceeds under mild reaction conditions and is tolerant to a large number of functional groups.19 Moreover, it is a click reaction that does not require metal catalyst in comparison to the copper-catalysed azide–alkyne click reaction which is an advantage for biological applications.20 In addition, our aim was to introduce an end-group compatible with other reactive groups incorporated along the polymer chains by copolymerization, allowing the synthesis of multifunctional polymers in a single copolymerization step. Those functional groups were chosen for their ability to react with primary-amine-functionalized substrates under mild conditions, without interference with the α-end-group. They were chloromethyl (from vinylbenzyl chloride) and N-succinimidyl ester (from N-succinimidyl acrylate).
Experimental section
Materials
Styrene (S, Fluka, ≥99%) and n-butyl acrylate (BA, Acros, >99%) were distilled under reduced pressure at room temperature before use. Vinylbenzyl chloride (VBC, Aldrich, 97%) was passed through a column of basic alumina to remove inhibitors. BlocBuilder® was kindly provided by Arkema (France). Dimethylformamide (DMF, VWR, Normapur), NaOH (ABCR, 99%), 3-butenylamine hydrochloride (Aldrich, 97%), benzyl mercaptan (ABCR, 99%), benzophenone (Aldrich, +99%) were used as received. The 2-methyl-2-[N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)-aminoxy]-N-propionyloxysuccinimide alkoxyamine was prepared according to the procedure reported by Vinas et al.11N-Succinimidyl acrylate (NSA) was synthesized as previously described.21
Analytical techniques
Conversions of the monomers for the copolymerizations and purity of the alkene-functionalized SG1-based alkoxyamine were determined by 1H NMR spectroscopy in CDCl3 at room temperature (using an AC250 Bruker spectrometer). Size exclusion chromatography (SEC) was performed in tetrahydrofuran (THF). Samples were analyzed at a concentration of 5 mg mL−1 after filtration through a 0.45 µm pore-size membrane. The flow rate was 1 mL min−1 and the separation was performed at 40 °C with two columns (PLgel Mixed-C, 7.5 mm × 300 mm; bead diameter: 5 µm; separation limits: 400 to 106 g mol−1). A differential refractive index detector (Viscotek VE 3580 RI Detector) was used, and molar masses (Mn, the number-average molar mass, Mw, the weight-average molar mass) and polydispersity indexes (PDI = Mw/Mn) were derived from a calibration curve based on polystyrene (PS) standards from Polymer Standards Service. In all plots showing the evolution of Mn with monomer conversion, the straight line corresponds to the expected evolution of the theoretical, Mn,th, calculated by the ratio of the initial mass of monomer multiplied by the conversion over the initial mole number of the alkoxyamine plus the molar mass of the latter. Mass spectra were recorded by matrix-assisted laser desorption and ionization time-of-flight (MALDI-TOF) mass spectrometry using a Bruker autoflex III smartbeam mass spectrometer, equipped with the laser that produces pulses at 337 nm using dithranol as a matrix and NaI as a cationizing agent. Spectra were recorded in linear mode and in reflector mode at accelerating potentials of 20 kV and 19 kV respectively. Samples were prepared by dissolving the polymer in THF at a concentration of 2–5 mg mL−1. A 10 µL aliquot of this solution was mixed with 20 µL of matrix solution and 10 µL of NaI solution (both at 20 mg mL−1 in THF). Standards (polystyrenes of known structure, Mn = 1500 and 4800 g mol−1 purchased from Polymer Standards Service) were used to calibrate the mass scale. Simulated mass spectrum was obtained using the IsoPro 3.0 program. In all cases, to determine m/z, the molar mass of the sodium cation was added.
Synthesis of the alkene-functionalized SG1-based alkoxyamine (MAMA–ene)
3-Butenylamine hydrochloride (0.317 g; 2.94 mmol) was dissolved in DMF (6.1 g) in the presence of NaOH (0.66 mL of NaOH 5 M in water). After stirring at room temperature for 3 min, 1 g of MAMA–NHS (2.1 mmol) and 3 g of DMF were added. The solution was deoxygenated by nitrogen bubbling for 15 min. After stirring at room temperature for 26 h, the solution was filtrated and the solvents were evaporated under high vacuum at room temperature to afford a crude product which was dissolved in a mixture of pentane (14 mL) and water (7 mL). The organic solution was extracted and pentane was removed in vacuum, affording the product as a slightly yellow oil (0.55 g, yield = 70%). 1H NMR (CDCl3), δ (ppm from TMS): 1.08–1.35 (m, 24H), 1.53 (s, 3H), 1.67 (s, 3H), 2.25 (m, 2H), 3.00–3.35 (m, 2H), 3.30 (d, J(H,P) = 26 Hz, 1H), 3.90–4.33 (m, 4H), 5.0 (m, 2H), 5.75 (m, 1H), 8.08 (s, 1H). Electrospray mass spectrometry performed on MAMA–ene (C21H43N2O5P, M = 434.5 g mol−1) revealed a peak at m/z 457.28 for [M + Na]+. Elemental analysis: calculated for C21H43N2O5P (M = 434.5 g mol−1): C, 58.04; H, 9.97; N, 6.45. Found: C, 57.45; H, 9.44; N, 6.41%.
Nitroxide-mediated homopolymerization of S and BA
In a typical experiment (experiment P1, Table 1), a round-bottomed flask (25 mL) was loaded with styrene (6.79 g, 65 mmol), DMF (0.5 g) and MAMA–ene (0.126 g, 0.289 mmol). The mixture was deoxygenated by argon bubbling for 30 min in iced water and was then immersed in an oil bath thermostatted at 110 °C. Samples were periodically withdrawn via purged syringes to measure the conversion by gravimetry. After complete drying, the raw polymer from each sample was dissolved in THF for size exclusion chromatography analysis. After cooling, the polymer was recovered by precipitating its solution in MeOH at room temperature.
| Expt |
Monomer(s) [M]/mol L−1 |
[MAMA–ene]0/mmol L−1 |
Time /min |
Conv. (wt%) |
M
n,th
/g mol−1 |
M
n,SEC
a/g mol−1 |
PDIb |
Final compositiong (molar ratio) |
|
Number-average molar mass, Mn, determined by size exclusion chromatography using polystyrene standards.
Polydispersity index, PDI = Mw/Mn.
After purification by precipitation in MeOH.
Polymerization in the presence of free SG1 (4.4 mol% for P3′ and P3″; 5.3 mol% for P4′ with respect to the alkoxyamine).
After purification by dialysis.
In toluene solution.
Determined by 1H NMR.
|
|
P1
|
S (8.1) |
36.1 |
66 |
12.4 |
3339 |
4310 |
1.22 |
100/0 |
| 115 |
21.1 |
5393 |
6120 |
1.17 |
| 209 |
34.9 |
8628 |
8970 |
1.12 |
| 319 |
48.6 |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 836 836 |
11240 |
1.14 |
| 365 |
50.8 |
12364 |
11980 |
1.17 |
| 12380c |
1.08c |
|
P1
′
|
S (8.1) |
36.2 |
54 |
9.4 |
2633 |
3400 |
1.30 |
100/0 |
| 4580c |
1.16c |
|
P2
|
S (7.2) + VBC (0.84) |
33.5 |
65 |
17.8 |
5081 |
4880 |
1.20 |
(S/VBC) 87.7/12.3 |
| 155 |
36.9 |
10064 |
9330 |
1.14 |
| 233 |
51.4 |
13835 |
12070 |
1.17 |
| 297 |
64.1 |
17144 |
14100 |
1.17 |
| 409 |
80.5 |
21439 |
16080 |
1.22 |
|
P3
f
|
BA (3.4) |
68.3 |
83 |
48.1 |
3516 |
3380 |
1.43 |
100/0 |
| 190 |
68.2 |
4806 |
4570 |
1.39 |
| 305 |
83.7 |
5801 |
5050 |
1.48 |
| 370 |
89.5 |
6168 |
5320 |
1.50 |
|
P3′
|
BA (6.1) |
23.4d |
16 |
6.9 |
2739 |
2480 |
1.71 |
100/0 |
| 41 |
12.9 |
4707 |
4260 |
1.62 |
| 141 |
30.1 |
10443 |
10000 |
1.35 |
| 255 |
45.6 |
15601 |
15240 |
1.27 |
| 303 |
50.3 |
17170 |
15740 |
1.34 |
|
P3″
|
BA (6.1) |
23.0d |
16 |
4.0 |
1784 |
1670 |
1.71 |
100/0 |
| 1360e |
1.70e |
|
P4
f
|
BA (3.1) + NSA (0.35) |
13.7 |
30 |
22.3 |
7841 |
7050 |
1.59 |
(BA/NSA) 88.8/11.2 |
| 75 |
32.8 |
11328 |
10140 |
1.54 |
| 140 |
46.2 |
15778 |
13190 |
1.54 |
| 322 |
68.0 |
23037 |
21360 |
1.48 |
|
P4′
|
BA (6.3) + NSA (0.72) |
26.1d |
83 |
20.9 |
7852 |
7585 |
1.46 |
(BA/NSA) 88.7/11.3 |
| 120 |
37.7 |
13794 |
12520 |
1.42 |
| 140 |
51.0 |
18515 |
16690 |
1.35 |
| 195 |
58.4 |
21160 |
20770 |
1.31 |
Nitroxide-mediated copolymerization of BA and NSA
In the experiment P4 (Table 1), a round-bottomed flask (50 mL) was charged with NSA (1.34 g, 7.95 mmol), BA (9.02 g, 70.4 mmol), DMF (1.0 g), MAMA–ene (135 mg, 0.31 mmol) and toluene (10.0 g). The mixture was deoxygenated by argon bubbling for 30 min in iced water and was then immersed in an oil bath thermostatted at 110 °C. Samples were periodically withdrawn to measure the conversion by 1H NMR by comparing the integrated areas of characteristic signals of monomers and DMF. After complete drying, the raw polymer from each sample was dissolved in THF for size exclusion chromatography analysis.
Nitroxide-mediated bulk polymerization of BA with PS–MAMA–ene macroalkoxyamine initiator
A mixture of BA (6.4 g, 50.0 mmol), SG1 (0.3 mg, 0.001 mmol) and PS–MAMA–ene (0.301 g, 0.024 mmol, Mn,SEC = 12380 g mol−1, PDI = 1.08, experiment P1 in Table 1) was deoxygenated by bubbling argon for 30 min in iced water and was then immersed for 7.2 h in an oil bath thermostatted at 110 °C.
Thiol–ene photoreaction
In a quartz reactor, the polymer (alkene chain-end functionalized poly(n-butyl acrylate)) (Mn,NMR = 1940 g mol−1, PDI = 1.7, experiment P3″ in Table 1), 3 equivalents of benzyl mercaptan ([thiol] = 0.5 M) and 0.1 equivalent of benzophenone were dissolved in DMF. The mixture was then exposed to UV light (Dymax 2000 Light Curing System) for 24 h. The thiol–ene reaction was followed by 1H NMR. The polymer was purified by dialysis in acetone and characterized by SEC in THF.
Results and discussion
Synthesis of the alkene-functionalized SG1-based alkoxyamine
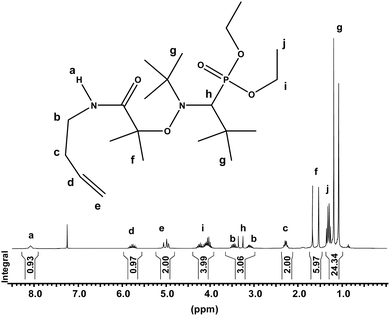
The new alkoxyamine initiator, MAMA–ene, bearing an alkene function was conveniently synthesized from MAMA–NHS and 3-butenylamine by amidification and further characterized (Scheme 1). The analytical data for MAMA–ene as presented in the experimental part were consistent with the proposed structure. As an illustration, Fig. 1 displays the 1H NMR spectrum recorded in CDCl3 at 25 °C showing the characteristic signals of the double bond in addition to those belonging to the SG1.
Nitroxide-mediated polymerizations
Non-reactive homopolymers, namely polystyrene, PS, and poly(n-butyl acrylate), PBA (P1 and P3, respectively) and their reactive counterparts, i.e. a copolymer of styrene and vinylbenzyl chloride, P(S-co-VBC) (P2) and a copolymer of n-butyl acrylate and N-succinimidyl acrylate, P(BA-co-NSA) (P4) were synthesized and the characterizations are reported in Table 1. As shown in Table 1 and illustrated in Fig. 2 for the experiment P1, all polymerizations were well-controlled as the recovered polymers exhibited a linear increase of Mn with monomer conversion along with polydispersity indexes below 1.7, even in the early stage of the polymerization. The number-average molar masses were in good agreement with the theoretical values indicating that the number of chains was governed by the alkoxyamine concentration and remained constant within the reaction times. Throughout the polymerization, SEC traces were symmetric and narrow (Fig. 2). Moreover, the reaction exhibited pseudo-first order kinetics as is required in controlled polymerization, indicating a constant concentration of propagating radicals and therefore a low contribution of irreversible termination reactions after establishment of the activation–deactivation equilibrium regulated by the persistent radical effect.22
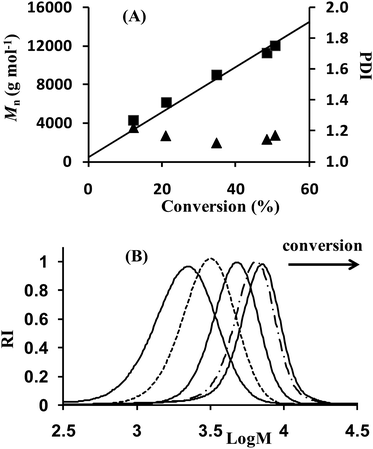 |
| | Fig. 2
Nitroxide mediated polymerization of styrene at 110 °C in the presence of MAMA–ene (expt P1 in Table 1). (A) Number average molar mass, Mn (square), and polydispersity index, Mw/Mn (triangle), determined by SECvs. conversion. The straight line corresponds to the theoretical Mnvs. conversion. (B) Size exclusion chromatograms as a function of monomer conversion. | |
The homopolymerization of n-butyl acrylate and its copolymerization with NSA (P3 and P4 in Table 1) in toluene solution led to well-defined polymers with, however, broader molar mass distribution than for the styrenic polymers. This situation cannot be ascribed to chain transfer to the polymer (via H-abstraction from the backbone methine) as low molar mass polymers were targeted and therefore the fraction of chains affected by transfer would remain low. It can rather be explained by chain transfer to the toluene. In order to improve livingness and achieve a better control, a small excess of free SG1 (about 5 mol% relative to the alkoxyamine concentration) was added and polymerizations were performed nearly in bulk (a small amount of DMF was used as an internal reference to determine monomer conversion by 1H NMR). Under these conditions, the number-average molar masses were closer to the theoretical values and the molar mass distributions were narrower (P3′ and P4′ in Table 1 and Fig. 3).
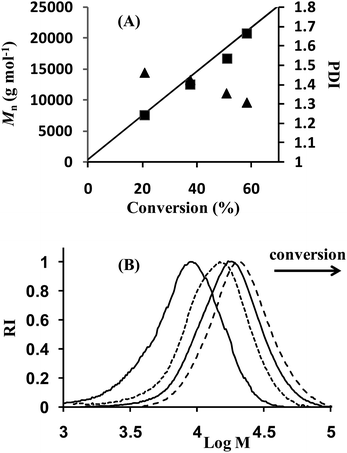 |
| | Fig. 3
Nitroxide mediated copolymerization of n-butyl acrylate with N-succinimidyl acrylate at 110 °C in the presence of MAMA–ene and 5.3 mol% of SG1 (expt P4′ in Table 1). (A) Number average molar mass, Mn (square), and polydispersity index, Mw/Mn (triangle), determined by SECvs. conversion. The straight line corresponds to the theoretical Mnvs. conversion. (B) Size exclusion chromatograms as a function of monomer conversion. | |
The good control over polymer characteristics is also an indication that potential chain transfer to the allylic proton was not an issue. To confirm this further, the proton NMR spectrum of a low molar mass poly(n-butyl acrylate) (P3″ in Table 1 and Fig. 4) showed the existence of both the vinyl protons (Hd and He) and the SG1-based alkoxyamine protons, both derived from the MAMA–ene initiator. On the basis of the intensity of the three vinylic protons of the alkoxyamine and the two ester protons of n-butyl acrylate units at 4 ppm, the calculated number-average molar mass was Mn,NMR = 1940 g mol−1, quite close to the value Mn,SEC = 1360 g mol−1 determined by SEC with PS calibration for this polymer.
The structure of one PS–MAMA–ene (P1′ in Table 1, Mn,SEC = 4580 g mol−1 and PDI = 1.16) was examined by MALDI-TOF mass spectrometry, in both linear and reflector modes, in order to confirm the presence of one initiator fragment containing the alkene group and one nitroxide (SG1) end group per chain. In the case of PS–MAMA–ene in linear mode, two main peak families can be observed with 104 mass units (the molar mass of the monomer unit) separating the peaks (Fig. 5).
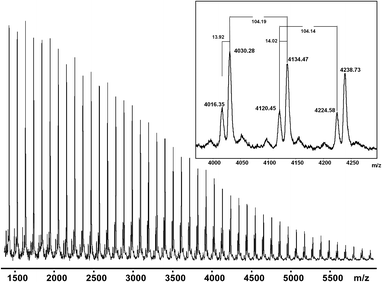 |
| | Fig. 5
MALDI-TOF mass spectra with NaI salt/dithranol matrix in linear mode (sum of 600 shots, in positive mode and with a laser repetition rate of 10.5 Hz) of polystyrene obtained by NMP initiated by MAMA–ene (experiment P1′). | |
The minor population corresponds to PS–MAMA–ene chains with loss of H-SG1 and the major one, which has a mass increment of +14 m/zvs. the minor one, corresponds to a scission of polystyrene chains induced by MALDI followed by a chain end-group degradation mechanism postulated by Schulte et al.23 during a study on a TEMPO–poly(N-isopropylacrylamide). Fragmentation during the MALDI-TOF MS analysis leading to polystyrene with a methylene end-group was also observed by other groups (Ladaviere et al.24 for a SG1–polystyrene, Dourges et al.25 for a TEMPO–polystyrene and Dempwolf et al.26 for TIPNO and N-tert-butyl-N-[1-isopropyl-2-methylpropyl] nitroxide (BIPNO)–polystyrene). With the reflector mode, only this latter population was observed (see the mass spectrum in the ESI, Fig. S1 and Table S1†). Consequently, under our MALDI experimental conditions, no peak series corresponding to a SG1-capped polystyrene was detected. Nevertheless, one can confirm the presence of alkene functionality located at the chain-end of the polymer chains. The molar mass values for the experimentally observed series are reported and compared with the theoretical values (average masses, Table 2). We also tried to use a more acidic 2,5-dihydroxybenzoic acid (DHB) matrix without any added salt in order to avoid polymer degradation during the MALDI-TOF MS analysis as suggested previously by Dourges et al.25 but this strategy failed with our SG1-capped polymer (see ESI, Fig. S2 and Table S2†).
Table 2 Comparison between the experimental and theoretical molar mass values of the Na+-ionized polystyrene obtained from MAMA–ene (dithranol/NaI matrix in linear mode)
| |
n
|
Calcd mass |
Expt mass |
Formula |
| Major series |
37 |
4029.84 |
4030.28 |
CH2CH–(CH2)2–NHCO–C(CH3)2–(styrene)n−1–CH2C(C6H5)CH2, Na+ |
| 38 |
4133.99 |
4134.47 |
| 39 |
4238.15 |
4238.73 |
| Minor series |
37 |
4015.82 |
4016.36 |
CH2CH–(CH2)2–NHCO–C(CH3)2–(styrene)n−1–CHCH–C6H5, Na+ |
| 38 |
4119.97 |
4120.45 |
| 39 |
4224.12 |
4224.58 |
The presence of the SG1 functionality at the chain-end of the polymer chains allows the insertion of a second monomer at this site to form an AB-type diblock copolymer. As an illustration, we have synthesized polystyrene-b-poly(n-butyl acrylate) from PS–MAMA–ene (P1 in Table 1, Mn,SEC = 12380 g mol−1 and PDI = 1.08). The size exclusion chromatogram of the block copolymer was narrow (Fig. 6) showing a very slight tailing towards the lower molar mass side. The number-average molar mass and polydispersity index of PS-b-PBA–MAMA–ene were determined by SEC (THF) as Mn = 74050 g mol−1 and PDI = 1.45. (The monomer conversion was determined to be 28.3% and theoretical Mn = 86800 g mol−1.) Thus, these results indicate a good overall efficiency of the PS–MAMA–ene macroalkoxyamine. Moreover, the 1H NMR spectrum of the block polymer clearly revealed the presence of both polymer blocks.
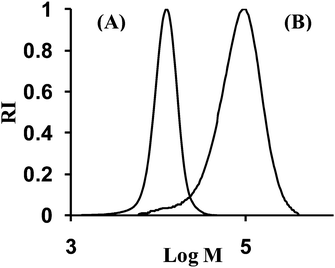 |
| | Fig. 6
SEC traces for: (A) PS–MAMA–ene P1 (Mn,SEC = 12380 g mol−1 and PDI = 1.08) prepared in toluene solution at 110 °C using MAMA–ene as the initiator and (B) polystyrene-b-poly(n-butyl acrylate) (Mn,SEC = 74050 g mol−1 and PDI = 1.45) prepared by chain extension of PS–MAMA–ene P1 in bulk with n-butyl acrylate at 110 °C. | |
Thiol–ene coupling reaction
Having shown that the new alkoxyamine is suitable to control the polymerization of acrylic and styrenic monomers and to produce block copolymers, we decided to demonstrate that the alkene functionality located at the chain-end of the polymer chains can be used as a reactive site for further chemical modification. In this context, we opted for the thiol–ene coupling reaction between benzyl mercaptan which absorbs at 254 nm and an alkene chain-end functionalized poly(n-butyl acrylate) (P3″ in Table 1, Mn,SEC = 1360 g mol−1 and PDI = 1.7) which does not absorb at this wavelength. We performed the experiment under photochemical conditions27 in the presence of benzophenone (10 mol%) as a photoinitiator. Monitored by NMR analysis (Fig. 7), the reaction was quantitative under these conditions. (The yield of functionalization determined by NMR was close to 100%, see ESI, Fig. S3†.) Indeed, the characteristic signals of the double bond of the alkene chain-end functionalized poly(n-butyl acrylate) disappeared in the 1H NMR spectrum of the crude reaction mixture. Moreover, in order to confirm the presence of the benzyl group at the end of poly(n-butyl acrylate) chains, we analyzed them in SEC by using a UV detector at 254 nm. Contrary to the alkene chain-end functionalized poly(n-butyl acrylate) used, the post-modified polymer was UV active (Fig. 8). Thus, these results indicate a good efficiency of the thiol–ene coupling reaction. The size exclusion chromatogram of the modified polymer was slightly shifted towards the higher molar masses mainly due to the dialysis step performed at the end of the thiol–ene reaction to remove the excess of mercaptan.
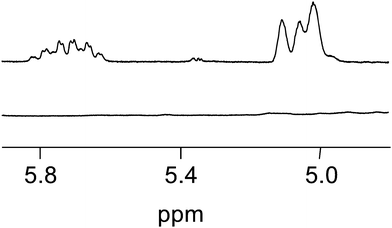 |
| | Fig. 7 Enlarged region of 1H NMR spectra of the alkene chain-end functionalized poly(n-butyl acrylate) (top) and the product after the thiol–ene coupling with benzyl mercaptan (bottom). | |
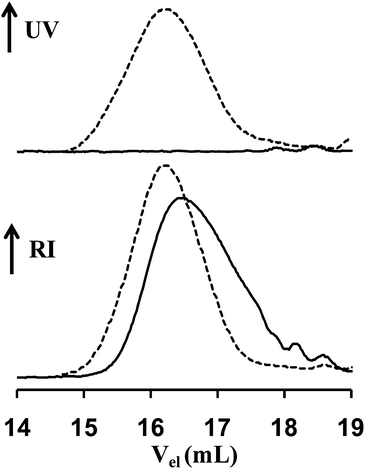 |
| | Fig. 8
SEC chromatograms obtained with a UV detector at 254 nm (top) and a refractive index detector (bottom) of the alkene chain-end functionalized poly(n-butyl acrylate) (full curve) and the product after the thiol–ene coupling with benzyl mercaptan (dashed curve). | |
Conclusions
A new alkene-functionalized SG1-based alkoxyamine has been synthesized in very simple conditions and used in order to prepare alkene chain-end functionalized polymers by NMP. The presence of the SG1 functionality at the ω-chain-end of the polymer chains allowed the insertion of a second monomer at this site to form an AB-type diblock copolymer. The alkene functionality located at the α-chain-end allowed the post-modification of the polymer by thiol–ene coupling reaction. The alkoxyamine was compatible with the polymerization of functional monomers, demonstrating its versatility toward the design of multifunctional copolymers of various architectures.
Acknowledgements
The French Ministry of Research and the CNRS are gratefully acknowledged for financial support of PhD student C.B. The authors also thank Arkema for kindly providing the BlocBuilder® alkoxyamine and Joël Belleney for his help in MALDI-TOF MS.
References
-
K. Matyjaszewki and T. P. Davis, Handbook of Radical Polymerization, Wiley, New York, 2002 Search PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- F. Chauvin, P.-E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. A. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238 CrossRef CAS.
- T. N. T. Phan and D. Bertin, Macromolecules, 2008, 41, 1886 CrossRef CAS.
- T. M. Eggenhuisen, C. R. Becer, M. W. M. Fijten, R. Eckardt, R. Hoogenboom and U. S. Schubert, Macromolecules, 2008, 41, 5132 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485 CrossRef CAS.
- B. Lessard and M. Maric, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2574 CrossRef CAS.
- S. R. S. Ting, E.-H. Min, P. Escale, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422 CrossRef CAS.
-
J.-L. Couturier and O. Guerret, Eur. Pat. Appl., EP 1464648, 2004.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Tetrahedron, 2005, 61, 8752 CrossRef CAS.
- J. Vinas, N. Chagneux, D. Gigmes, T. Trimaille, A. Favier and D. Bertin, Polymer, 2008, 49, 3639 CrossRef CAS.
- J. Parvole, L. Ahrens, H. Blas, J. Vinas, C. Boissiere, C. Sanchez, M. Save and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 173 CrossRef CAS.
- C. Chevigny, D. Gigmes, D. Bertin, J. Jestin and F. Boué, Soft Matter, 2009, 5, 3741 RSC.
- J. Ma, C. Cheng, G. Sun and K. L. Wooley, Macromolecules, 2008, 41, 9080 CrossRef CAS.
- K. Matyjaszewki, K. L. Beers, A. Kern and S. G. Gaynor, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 823 CrossRef CAS.
- F. Zeng, Y. Shen, S. Zhu and R. Pelton, Macromolecules, 2000, 33, 1628 CrossRef CAS.
- J. Ding, C. Chuy and S. Holdcroft, Macromolecules, 2002, 35, 1348 CrossRef CAS.
- D. Kuckling and S. Wohlrab, Polymer, 2001, 43, 1533.
- K. I. Rhiannon, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS.
-
(a) A. Pollak, H. Blumenfeld, M. Wax, R. L. Baughn and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 6324 CrossRef CAS;
(b) U. Heimgartner, B. Kozulic and K. Mosbach, Anal. Biochem., 1989, 181, 182 CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef CAS.
- T. Schulte, K. O. Siegenthaler, H. Luftmann, M. Letzel and A. Studer, Macromolecules, 2005, 38, 6833 CrossRef CAS.
- C. Ladaviere, P. Lacroix-Desmazes and F. Delolme, Macromolecules, 2009, 42, 70 CrossRef CAS.
- M. A. Dourges, B. Charleux, J.-P. Vairon, J.-C. Blais, G. Bolbach and J.-C. Tabet, Macromolecules, 1999, 32, 2495 CrossRef.
- W. Dempwolf, S. Flakus and G. Schmidt-Naake, Macromol. Symp., 2009, 275–276, 166 CrossRef CAS.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem.–Eur. J., 2009, 15, 11444 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.