Formation of nanoporous materials via mild retro-Diels–Alder chemistry†
Mathias
Glassner
,
James P.
Blinco
and
Christopher
Barner-Kowollik
*
Preparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128, Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; Web: http://www.macroarc.de Fax: +49 721 608 5740
First published on 4th October 2010
Abstract
Poly(styrene)-block-poly(ethylene oxide) copolymers synthesized via the combination of reversible addition fragmentation chain transfer (RAFT) polymerization and hetero Diels–Alder (HDA) cycloaddition can be cleaved in the solid state by a retro-HDA reaction occurring at 90 °C. Nanoporous films can be prepared from these polymers using a simple heating and washing procedure.
Introduction
Materials which contain nanoporous architectures have received remarkable attention because of their attractive applications as membranes, sensors and in catalysis.1,2 One technique for the generation of nanoporous materials is to commence with an amphiphilic block copolymer (BCP) which undergoes phase separation and to subsequently remove one block preferentially.Currently, the most common methodology employed for this removal is the destruction of one block via ozonolysis,3,4 UV degradation,5–7reactive ion etching8 or chemical etching.9–14 However, these techniques are limited to few systems as several polymers undergo side chain hydrolysis or degradation under such harsh conditions. A strategy that avoids possible degradation problems is the introduction of a labile linker between two blocks with differing solubilities that can be cleaved under mild reaction conditions. Such an approach allows one block to be removed by a simple washing process once cleavage has taken place. Additionally, a judicious choice of juncture and cleavage conditions can produce pore walls that bear reactive functional groups which provide access to further chemical transformations.10,15 Penelle et al. prepared poly(styrene)-block-poly(methyl methacrylate) copolymers with a [4 + 4] anthracene photodimer at the junction that could be cleaved either photolytically or thermally.16 Poly(styrene)-block-poly(ethylene oxide) (PS-b-PEO) linked with a thermally labile alkoxyamine between the PS and PEO segments was also prepared, viafree radical polymerization of styrene utilizing a TEMPO functionalized PEO block.17PS-b-PEO is the most frequently used block copolymer for the preparation of nanoporous thin films, because simple solvent-annealing leads to highly ordered arrays of PEO cylinders oriented normal to the film’s surface.18PS-b-PEO block copolymers with various linkers have been employed for the construction of nanoporous thin films including an acid-sensitive trityl ether,19 a photo-cleavable ortho-nitrobenzyl moiety,20 and a disulfide bond that undergoes scission upon exposure to a redox stimuli.21
A simple approach for the synthesis of various block copolymers is the conjugation of end-group functionalized macromolecular blocks via efficient coupling reactions.22 One reaction that has been used for such coupling is the hetero Diels–Alder (HDA) reaction between a diene end-functionalized polymer and the electron deficient dithioester end-group of a polymer synthesized by reversible addition fragmentation chain transfer (RAFT) polymerization.23–25Copolymers formed via the RAFT-HDA methodology have the added advantage that the thiopyran linkage has been shown to be reversible, undergoing a retro-Diels–Alder (r-DA) reaction upon thermal treatment in solution.24,26 However, the open-chain diene employed in ref. 26 required temperatures above 120 °C at which point the released dithioesters were unstable and underwent rapid degradation. Recently, it was shown that the use of more reactive cyclopentadienyl functional polymers also resulted in the reduction in the required temperature (ca. 80–90 °C) for the r-DA reaction to take place.27
Herein, we describe the synthesis of PS-b-PEO copolymersvia the RAFT-HDA approach which can subsequently undergo a quantitative cleavage into the constituent blocks in the solid state under mild heating conditions. We further demonstrate that nanoporous polystyrene films can be prepared from these copolymers after the r-DA reaction as the PEO segments can easily be removed by washing the films with water.
Results and discussion
The synthetic pathway for both blocks and the HDA conjugation is depicted in Scheme 1. Cyclopentadienyl (Cp) functionalized PEO 1 was synthesized by tosylation of monomethylated PEO followed by conversion into the Cp-functionalized polymer using the recently reported nickelocene method.28Benzylpyridin-2-yldithioformate was used as controlling agent to prepare poly(styrene) 2a,b by RAFT polymerization. The RAFT-HDA cycloaddition of PEO 1 and PS 2a,b was performed in chloroform in the presence of trifluoroacetic acid (TFA). | ||
| Scheme 1 Synthetic strategy for producing cleavable PS-b-PEOvia the RAFT-HDA click reaction. The reaction conditions are as follows: (a) TsCl, NaOH, THF/H2O; (b) NiCp2, NaI, PPh3, THF, 40 °C; and (c) RAFT polymerization, AIBN, 60 °C. | ||
Fig. 1 shows an overlay of SEC traces of the individual building blocks PEO 1 and PS 2a,b as well as the coupling product 3a,b. A clear shift of the distributions to lower retention times indicates the successful formation of PS-b-PEO which is consistent with the estimated number average molecular weights, Mn. To investigate the thermal cleavage of the block copolymer in the solid state, PS-b-PEO (3b) was suspended in water and heated to 90 °C. The insoluble residue was collected by filtration at predetermined time intervals (4–30 h) and analyzed viaSEC, as shown in Fig. 2a. Inspection of Fig. 2a indicates that after just 4 h of heating the majority of the BCP has undergone a r-DA with the disappearance of the SEC elugram of 3b and the reappearance of the elugram corresponding to the homopolymer block 2b. The SEC elugram of 1 is not observed as once the r-DA has occurred it dissolves in the water and is removed during filtration. Under these conditions the r-DA reaction was essentially quantitative after 20 h. 1H-NMR analysis of the recovered solid, after precipitation in methanol, also demonstrated that the majority of the PEO was removed, as can be seen from the decrease of the PEO backbone signal at 3.62 ppm (Fig. 2b). A comparison of the integral ratios shows that only 10% of 3b is remaining. The data from Fig. 2 and the fact that the HDA reaction between a Cp-functionalized polymer and the pure RAFT agent is fully reversible27 allowed for the conclusion that block copolymers with a shorter PS block such as 3a can undergo a complete r-DA reaction as well.
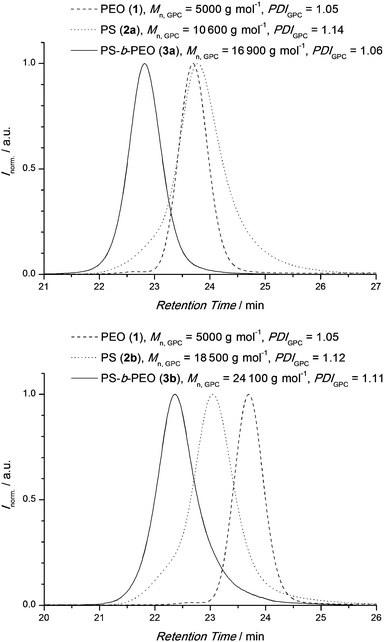 | ||
| Fig. 1 Overlay of SEC traces showing the formation of PS-b-PEO 3a (above) and 3b (below). The BCPs were purified by extraction of residual homo-PS with diethyl ether and precipitation in methanol. | ||
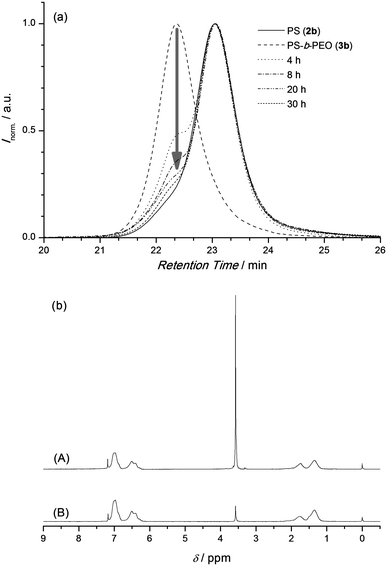 | ||
| Fig. 2 (a) SEC monitoring of the r-DA cleavage of PS-b-PEO (3b, 18.5-b-5.0 kg mol−1) in the solid state in water at 90 °C. (b) 1H-NMR in CDCl3 of PS-b-PEO (A) and of the precipitate in MeOH after 20 h at 90 °C (B). | ||
The fact that dithioesters are colored due to a π → π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond can be used for a direct monitoring of the r-DA reaction by UV-Vis spectroscopy because the absorbance is proportional to the concentration of the dithioester capped PS.
S double bond can be used for a direct monitoring of the r-DA reaction by UV-Vis spectroscopy because the absorbance is proportional to the concentration of the dithioester capped PS.
UV-Vis absorbance spectra of PS-b-PEO (3b) in toluene (0.7 mmol L−1) at 90 °C are depicted in Fig. 3. One can observe an increase in the absorbance with time due to an increasing concentration of the dithioester functionalized PS (2b). The maximum absorbance occurs at the same wavelength (521 nm) as for the pure RAFT polymer (see Fig. S1 in the ESI†). After 120 min, no further increase of the absorbance was observed which indicates a faster reaction in solution compared to the solid state.
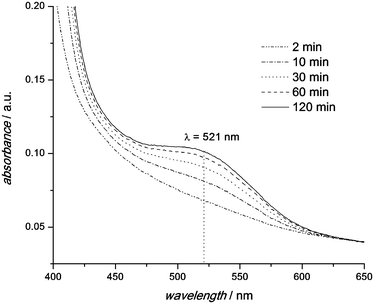 | ||
| Fig. 3 UV-Vis spectra of PS-b-PEO (3b) in toluene at 90 °C. Due to the r-DA reaction the concentration of the dithioester capped PS (2b) increases. | ||
Once optimum cleavage conditions were established for PS-b-PEO, an investigation into the removal of the PEO block from a thin film by washing with water after the r-DA reaction was undertaken. PS-b-PEO films were drop-cast on silicon wafers from a 2 wt% solution in chloroform. After drying under vacuum, the films were heated at 90 °C for 15 h, washed with water to remove the PEO and dried again under vacuum.
Scanning electron microscopy (SEM) images of PS-b-PEO films after heating and washing with water are displayed in Fig. 4. The formation of random nanopores can be clearly seen in Fig. 4a which proves that the PEO block could be removed by the simple heating and washing procedure. SEM images of various blind samples (PS-b-PEO 3a,b before heating, PS 2b after heating/washing and PS-b-PEO 3b after washing with water) showing no pore formation are displayed in Fig. S2 (see the ESI†). While a porous structure is formed, compared with other nanoporous systems,9–15,19–21 employing PS-b-PEO 3b (18.5-b-5.0 kg mol−1) only yielded a film with a limited number of pores. When considering the size of the blocks used to form 3b, PEO only accounts for close to 20 wt% of the total polymer system. It is thus not surprising that only few pores are formed. Increasing the fraction of PEO within the block copolymer should lead to an increase in the number of pores formed in the material. To achieve a pore count increase, a shorter PS block was employed in the initial RAFT-HDA reaction forming PS-b-PEO 3a (10.6-b-5.0 kg mol−1) in which the overall percentage of PEO within the BCP was close to 50 wt%. When 3a undergoes the same r-DA treatment and washing as 3b, a marked increase in porosity of the film can be observed (see Fig. 4b). As depicted in Fig. 4b (and more clearly in the enlargement, Fig. 4c) the pore diameters range from 40 nm to 200 nm, which is in the same size range as the pores observed in Fig. 4a, yet there is an over six-fold increase in the number of observed pores. Importantly, the cross-section SEM image of a freeze-fractured sample in Fig. 4d confirms that the pore formation is not limited to the film's surface but occurs throughout the entire bulk of the film. As the PEO block is synthesized independently to the PS block, interchanging different PS blocks can be achieved with ease. Such an approach should allow the described technique to be extended further, enabling the tuning of film porosity by substituting PS blocks of differing lengths.
 | ||
| Fig. 4 SEM images of (a) PS-b-PEO 3b (18.5-b-5.0 kg mol−1) after heating to 90 °C and washing with water; (b) PS-b-PEO 3a (10.6-b-5.0 kg mol−1) after heating to 90 °C and washing with water; (c) PS-b-PEO 3a (10.6-b-5.0 kg mol−1) after heating to 90 °C and washing with water; and (d) freeze-fracture cross-section of PS-b-PEO 3a (10.6-b-5.0 kg mol−1) after heating to 90 °C and washing with water. | ||
In summary, it was possible to synthesize thermally cleavable PS-b-PEO block copolymers by RAFT-HDA click chemistry. The cleavage of PS-b-PEO can be carried out by heating to 90 °C in the solid state (or in solution). UV-Vis analysis confirmed that the dithioester end-group of the RAFT polymer is returned to the reformed homopolymer and that it is stable under these conditions. Nanoporous PS films were readily prepared by removal of the PEO block by applying a simple heating and washing procedure. The method described here enables a facile preparation of nanoporous films bearing a reactive thiocarbonyl thio group which provides access to further transformations from block copolymer precursors which are accessible in various molecular weight compositions via the combination of RAFT polymerization and HDA cycloaddition.
Experimental
Detailed synthetic procedures and characterization data for all polymers are given in the ESI†.Film casting
PS-b-PEO films were drop-cast onto silicon wafers from a 2 wt% solution in chloroform. After drying under vacuum, the films were heated at 90 °C for 15 h, washed with water to remove the PEO and dried again under vacuum. The cross-section was prepared by fracturing the sample immediately after submerging in liquid nitrogen.Measurements
The structures of the synthesized compounds were confirmed by 1H-NMR spectroscopy using a Bruker AM 400 spectrometer at 400 MHz for hydrogen nuclei. All samples were dissolved in CDCl3. Size exclusion chromatography (SEC) measurements were performed on a Varian PL50 system, comprising an auto injector, a Polymer Laboratories 5.0 µm bead-size guard column, followed by three linear PL columns (PLgel 5 µm MIXED-C) and a differential refractive index detector using dimethylacetamide (DMAc) as the eluent at 50 °C with a flow rate of 1 mL min−1. The SEC system was calibrated using narrow polystyrene standards ranging from 160 to 6 × 106 g mol−1. UV-Vis spectra were recorded on a Varian Cary 300 Bio spectrophotometer. 50 mg polymer was dissolved in 3 mL of toluene, filtered and placed in a UV-Vis cuvette within a heatable sample cell holder. Scanning electron microscopy (SEM) was performed on a FEI Quanta 650 FEG ESEM operating at 5.0 kV accelerating voltage.Acknowledgements
C.B.-K. acknowledges financial support from the Karlsruhe Institute of Technology (KIT) in the context of the Excellence Initiative for leading German universities as well as the German Research Council (DFG) and the Ministry of Science and Arts of the state of Baden-Württemberg. M.G. acknowledges financial support from the Landesgraduiertenförderung Baden-Württemberg. J.P.B. acknowledges funding by the Alexander von Humboldt Foundation (Bonn, Germany; Humboldt Research Fellowship). We also gratefully acknowledge Dipl. Ing. Volker Zibat (KIT, Laboratory for Electron Microscopy) for performing the SEM measurements.References
- M. A. Hillmyer, Adv. Polym. Sci., 2005, 190, 137–181 CAS.
- D. A. Olson, L. Chen and M. A. Hillmyer, Chem. Mater., 2007, 20, 869–890.
- P. Mansky, C. K. Harrison, P. M. Chaikin, R. A. Register and N. Yao, Appl. Phys. Lett., 1996, 68, 2586–2588 CrossRef CAS.
- J. S. Lee, A. Hirao and S. Nakahama, Macromolecules, 1988, 21, 274–276 CrossRef CAS.
- T. Thurn-Albrecht, R. Steiner, J. DeRouchey, C. M. Stafford, E. Huang, M. Bal, M. Tuominen, C. J. Hawker and T. P. Russell, Adv. Mater., 2000, 12, 787–791 CrossRef CAS.
- U. Jeong, H.-C. Kim, R. L. Rodriguez, I. Y. Tsai, C. M. Stafford, J. K. Kim, C. J. Hawker and T. P. Russell, Adv. Mater., 2002, 14, 274–276 CrossRef CAS.
- J. Bang, S. H. Kim, E. Drockenmuller, M. J. Misner, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2006, 128, 7622–7629 CrossRef CAS.
- R. Olayo-Valles, M. S. Lund, C. Leighton and M. A. Hillmyer, J. Mater. Chem., 2004, 14, 2729–2731 RSC.
- J. M. Leiston-Belanger, T. P. Russell, E. Drockenmuller and C. J. Hawker, Macromolecules, 2005, 38, 7676–7683 CrossRef CAS.
- A. S. Zalusky, R. Olayo-Valles, J. H. Wolf and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 12761–12773 CrossRef CAS.
- J. Rzayev and M. A. Hillmyer, Macromolecules, 2004, 38, 3–5.
- H. Mao and M. A. Hillmyer, Macromolecules, 2005, 38, 4038–4039 CrossRef CAS.
- B. W. Boudouris, C. D. Frisbie and M. A. Hillmyer, Macromolecules, 2007, 41, 67–75.
- L. M. Pitet, M. A. Amendt and M. A. Hillmyer, J. Am. Chem. Soc., 2010, 132, 8230–8231 CrossRef CAS.
- J. Rzayev and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 13373–13379 CrossRef CAS.
- J. T. Goldbach, T. P. Russell and J. Penelle, Macromolecules, 2002, 35, 4271–4276 CrossRef CAS.
- F. J. Hua and Y. L. Yang, Polymer, 2001, 42, 1361–1368 CrossRef CAS.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226–231 CrossRef CAS.
- M. Zhang, L. Yang, S. Yurt, M. J. Misner, J.-T. Chen, E. B. Coughlin, D. Venkataraman and T. P. Russell, Adv. Mater., 2007, 19, 1571–1576 CrossRef CAS.
- M. Kang and B. Moon, Macromolecules, 2008, 42, 455–458 CrossRef.
- J.-H. Ryu, S. Park, B. Kim, A. Klaikherd, T. P. Russell and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 9870–9871 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120–4126 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CAS.
- S. Sinnwell, C. V. Synatschke, T. Junkers, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2008, 41, 7904–7912 CrossRef CAS.
- T. Paulöhrl, A. J. Inglis and C. Barner-Kowollik, Adv. Mater., 2010, 22, 2788–2791 CrossRef.
- A. J. Inglis, T. Paulöhrl and C. Barner-Kowollik, Macromolecules, 2009, 43, 33–36.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization data for all polymers, UV-Vis spectrum of RAFT-PS, and SEM images of various blind samples. See DOI: 10.1039/c0py00267d |
| This journal is © The Royal Society of Chemistry 2011 |