Polymeric vesicles with well-defined poly(methyl methacrylate) (PMMA) brushes via surface-initiated photopolymerization (SIPP)
Fengting
Chen
a,
Xuesong
Jiang
*a,
Rui
Liu
b and
Jie
Yin
*a
aSchool of Chemistry & Chemical Technology, the State Key Lab. of Metal Matrix Composites, Shanghai Jiao Tong University, Shanghai, 200240, People's Republic of China. E-mail: ponygle@sjtu.edu.cn; Fax: +86-21-54747445; Tel: +86-21-54743268jyin@sjtu.edu.cn
bNational Engineering Research Center for Nanotechnology, Shanghai, 200240, People's Republic of China
First published on 5th November 2010
Abstract
We describe a novel and general approach to fabricate polymeric vesicles with well-defined PMMA brushes by using silica particles as template through surface-initiated photopolymerization (SIPP). The crosslinked PDMAEMA layer was immobilized on the surface of silica particles, and then photoinitiated polymerization of methyl methacrylate (MMA) was used to generate PMMA brushes in the presence of thioxanthone (TX). Polymeric vesicles with well-defined PMMA brush were obtained after removing silica cores. The whole process was well traced and confirmed by TEM, SEM, FT-IR and TGA. To the best of our knowledge, this is the first report the fabrication of polymer brush on the surface of polymeric vesicles through SIPP. This robust approach is expected to have potential in fabrication and modification of polymer vesicles with different sizes and functions.
Introduction
Construction of polymeric vesicles is of great interest because of their potential applications in encapsulators for controlled release of drugs, enzymes, dyes, paints1–5 and protection of active species.6 Much effort has been made to fabricate polymer vesicles with many novel and useful structures. A variety of methods, consisting mainly of a sacrificial core method,7–10self-assembly of amphiphlic block copolymers11,12 and all sorts of emulsion polymerizations13–15 have been developed for preparing polymeric vesicles. As one of the important methods for the preparation of polymer vesicles, silica particles are usually employed as sacrificial templates because of their availability in wide range of uniform-sizes.16,17 Through dispersion polymerization, Xu and Asher prepared monodisperse polymer shell-SiO2 core particles and removed core by HF etching to produce hollow polymer particles.9 By using layer-by-layer self-assembly technology, Connal et al. fabricated polymer vesicles through depositing PMAA and PVP on silica particle templates.18 Recent development in surface-initiated polymerization directly on silica particle template has provided an alternative approach to the preparation of inorganic/organic core-shell hybrids with well-defined and controlled shell structures and the corresponding polymeric vesicles by removal of the core.10,19 Fukuda and coworkers prepared hollow spheres from mono-dispersed silica particles grafted with oxetane-holding polymer brushes by the ring-opening reactivity of oxetane for shell cross-linking.10Due to their potential application in biomaterials, functionalization and surface-modification of polymeric vesicles became very important, as this allows tuning of the polymeric vesicles' properties for different purposes. As a very powerful alternative to modify the surface with molecular precision, polymer brushes have been attracting much attention because of their potential applications in many surface-based technologies such as biomaterials, adhesion and wetting, molecular recognition, microfluidics and nano-sensors.20–24 Quite a few methods for SIP have been used to fabricate polymer brushes on a variety of substrates, in particular photopolymerization and controlled polymerizations.25–27 Among the various polymerizations, photopolymerization has been widely used as a simple, versatile and effective approach to synthesize polymer brush with high grafting density and uniformed thickness.28–30 Dyer and coworkers have been dedicated to synthesize polymer brush by photopolymerization and they have reported grafting densities as high as 1.4 chains nm−2 for polystyrene brushes on gold.28 And we also reported our latest achievement of fabricating well-defined PMMA brushes on silica particles by SIPP. The thickness of PMMA brush can reach 25 nm upon exposure to UV light for five hours.30
Herein, we immobilized crosslinked PDMAEMA shell containing coinitator amino group on silica particles' surfaces, which can initiate MMA in the presence of photoinitiator TX under UV-irradiation. Polymeric vesicles with well-defined PMMA brushes can easily be obtained after removing the silica cores by HF. It is noteworthy that this technique is easy and robust for the synthesis of all kinds of vesicles by changing the monomer for brush formation. To the best of our knowledge, it is the first time to modify polymer vesicles with polymer brush through surface-initiated photopolymerization (SIPP).
Experimental section
Materials
(3-Mercaptopropyl)trimethoxysilane (MPS, 95%, Alfa Aesar), tetraethoxysilane (TEOS), ethanol, ammonia, N,N-dimethylformamide (DMF) and other solvents are purchased from Sinopharm Chemical Reagent Co., Ltd. (SCRC) and all of analytical grade without further purification. Methyl methacrylate (MMA, SCRC) and 2-(dimethylamino)ethyl methacrylate (DMAEMA, 98%, Sigma-Aldrich) was purified by washing with 5% NaOH to remove inhibitor and then double distillation under reduced pressure; it was then stored in the refrigerator before using. Anhydrous toluene was prepared by drying with anhydrous calcium hydride and then distillation under reduced pressure before using. Azobisisobutyronitrile (AIBN, 98%, SCRC) and N,N′-methylenebis(acrylamide) (MBA, 99%, SCRC) recrystallized in ethanol and acetone, respectively. 2-(2-3-Epoxypropyloxy) thioxanthone (ETX) was synthesized in our lab according to the procedure described in the literature.31Characterization
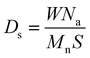 | (1) |
The cross-linked shells were synthesized via normal free radical reaction. Typically, 0.25 g of SiO2@MPS was redispersed in 25 mL of fresh DMF and 0.01 g of AIBN, 1 mL (6 mmol) of DMAEMA and 0.025 g (0.16 mmol) of MBA were added into the solution. The reaction was heated with constant stirring at 70 °C for 10 h under dry nitrogen and then cooled to room temperature. The core-shell particles (SiO2@PDMAEMA) were separated away from impurities and byproducts by centrifugation/redispersion for three times using DMF. At last, the hybrids were redispersed in 25 ml of fresh DMF.
Results and discussion
Immobilization of cross-linked PDMAEMA layer
As illustrated in Scheme 1, our strategy for synthesis of polymer vesicles with PMMA brushes involved three steps: immobilization of cross-linked PDMAEMA shell on SPs, surface-initiated photopolymerization of MMA and preparation of polymeric vesicles, which were well traced by TEM, SEM FT-IR and TGA. In this work, silica particles of 350 nm in diameter, as determined by TEM and SEM (Fig. 1a and 2a), were synthesized through the well-known Stöber process.32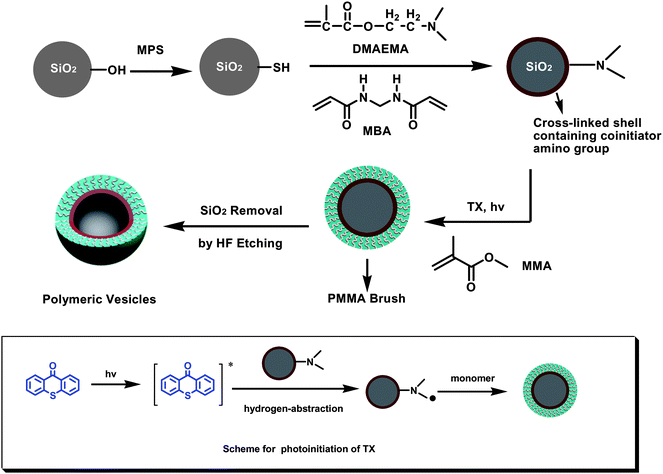 | ||
| Scheme 1 The process for synthesis of polymeric vesicles with well-defined PMMA brush. | ||
 | ||
| Fig. 1 TEM images of SiO2 (a), SiO2@MPS (b), SiO2@PDMAEMA (c), SiO2@PDMAEMA@PMMA-3h (d), SiO2@PDMAEMA@PMMA-7h (e), SiO2@PDMAEMA@PMMA-7h (magnified image) (f), PDMAEMA@PMMA-3h (g) and PDMAEMA@PMMA-7h (h). | ||
 | ||
| Fig. 2 SEM images of SiO2 (a), SiO2@MPS (b), SiO2@PDMAEMA (c), SiO2@PDMAEMA@PMMA-7h (d), PDMAEMA@PMMA-3h (e), and PDMAEMA@PMMA-7h (f). | ||
In the process of immobilization of the cross-linked PDMAEMA shell, thiol groups were firstly introduced to the surface of SPs by chemical modification of raw SPs using (3-mercaptopropyl)trimethoxysilane (MPS). The radical transfer addition polymerization took place between the thiol groups on the SPs surfaces and the propagating chains of PDMAEMA. In order to avoid inter-particle cross-linking, we did a series of experiments to find out the optimum conditions. We found that the particle concentration and the dosage of cross-linking agent are key factors to the success of this procedure. The particle concentration should be low and here we add 0.25 g silica particles into the 25 ml DMF solution (10 mg mL−1). In addition, the optimum dosage of crosslinking agent is 2.0 wt %–4.0 wt % (with respect to DMAEMA monomer).
To confirm that the cross-linked PDMAEMA layer was successfully fixed on the silica particles, TEM, SEM, FTIR and TGA traced the process. Fig. 1c shows a TEM image of the cross-linked shell of PDMAEMA with ∼5 nm thickness. Compared with the raw SPs (SiO2, Fig. 1a) and SiO2@MPS (Fig. 1b), there was an obvious organic layer in TEM image of SiO2@PDMAEMA, which was further confirmed by SEM (Fig. 2c), and FT-IR (Fig. 3). As comparison to FT-IR spectra of raw SPs (SiO2) and SiO2@MPS (Fig. 3a and 3b), the appearance of absorbance band at 1730 cm−1 in Fig. 3c, was characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations for PDMAEMA. The slightly bigger (dSiO2@PDMAEMA ≈ 360 nm) hybrid particles shown in the SEM image (Fig. 2c) also supported our conclusion. All these results are the best evidence to confirm the successful attachment of PDMAEMA layer on SPs. The amount of PDMAEMA on the surface of SPs can be further determined by TGA analysis. In Fig. 4, there exists 1.4% difference between raw SPs and SiO2@MPS, and 4.2% difference between SiO2@MPS and SiO2@PDMAEMA at 700 °C. We can roughly estimate that the graft density of MPS on SPs' surface is ∼5.22 chain/nm2. If the density of PDMEMA cross-linked layer is ∼1.0 g cm−3, the PDMAEMA layer thickness can be estimated to be ∼5.1 nm based on the TGA data, which is in good agreement with TEM and SEM results.
O vibrations for PDMAEMA. The slightly bigger (dSiO2@PDMAEMA ≈ 360 nm) hybrid particles shown in the SEM image (Fig. 2c) also supported our conclusion. All these results are the best evidence to confirm the successful attachment of PDMAEMA layer on SPs. The amount of PDMAEMA on the surface of SPs can be further determined by TGA analysis. In Fig. 4, there exists 1.4% difference between raw SPs and SiO2@MPS, and 4.2% difference between SiO2@MPS and SiO2@PDMAEMA at 700 °C. We can roughly estimate that the graft density of MPS on SPs' surface is ∼5.22 chain/nm2. If the density of PDMEMA cross-linked layer is ∼1.0 g cm−3, the PDMAEMA layer thickness can be estimated to be ∼5.1 nm based on the TGA data, which is in good agreement with TEM and SEM results.
 | ||
| Fig. 3 FT-IR of SiO2 (a), SiO2@MPS (b), SiO2@PDMAEMA (c), SiO2@PDMAEMA@PMMA (d) and PDMAEMA@ PMMA (e). | ||
 | ||
| Fig. 4 TGA of SiO2 (raw SPs) (a), SiO2@MPS (b), SiO2@PDMAEMA (c), SiO2@PDMAEMA@PMMA-3h (d) and SiO2@PDMAEMA@ PMMA-7h (e). | ||
Surface-initiated photopolymerization of MMA
Due to the abundant amino groups in the co-initiator PDMAEMA, the cross-linked PDMAEMA layer can generate radicals under UV-exposure to initiate photopolymerization of MMA in the presence of hydrogen-abstraction photoinitiator thioxanthone (TX). Due to the shield of light caused by particles (diameter 100 nm), it is a challenge to synthesize polymer brush on particles by photochemical approach. Because of the excellent UV-absorbance at 360–400 nm,33,34TX can well-absorb UV light to produce radicals even in the case of particles' shield and is consequently widely used in photocuring systems containing pigment particles such as coating and printing ink.35 Using TX as photoinitiator, we previously fabricated well-defined PMMA brushes directly on SP of 200 nm in diameter through SIPP.30 In the presence of co-initiator amino groups, the photolysis of thioxanthone can lead to formation of a active radical produced from TX (ketyl radical) and another radical derived from hydrogen donor amine. The polymerization of MMA is usually initiated by amino radicals, and the ketyl radicals are not active because of steric hindrance. Therefore, the PMMA brushes can be grafted on the surface of the PDMAEMA layer. The morphology of the obtained SiO2@PDMAEMA@PMMA hybrid particles were revealed by TEM (Fig. 1d, e and f). According to TEM images shown in Fig. 1, well-defined PMMA brush on the surface of cross-linked PDMAEMA was successfully synthesized through SIPP. With the increase of exposure time, the PMMA brush layer became obviously thicker (Fig. 1d and e), which indicated that the thickness of PMMA brush can be controlled by photopolymerization time. After photopolymerization for 7 h, the mean thickness of PMMA brush layers is about 45 nm and the diameter of SiO2@PDMAEMA@PMMA-7h is about 440 nm according to images of TEM and SEM (Fig. 1e, f and 2d). The formation of the PMMA brush layer can be further confirmed by FT-IR and TGA. Compared with FT-IR spectra of SiO2@PDMAEMA in Fig. 3c, the significant increase peak at 1730 cm−1 derived from CO of PMMA in Fig. 3d, indicating the successful attachment of PMMA brush on SPs. The amount of PMMA brush layer can be determined by TGA (Fig. 4), and the detailed amount of PMMA brush layer at different exposure time is summarized in Table 1. If the density of the PMMA organic layer is about 1.04 g cm−3, the organic layer thickness of SiO2@PMMA-7h can be estimated to be 45.5 nm based on the data of TGA, which is in good agreement with TEM result (∼45 nm). Due to chain transfer in the photopolymerization of MMA, the free PMMA can be generated in the solution besides PMMA brush attached on the surface of silica particles. The molecular weight (Mn) and polydispersity of the free PMMA in solution were checked by GPC, which can be considered to be the same to those of PMMA brush on the substrate.36–38 The Mn of PMMA is about 7.2 × 104 for first 3 h of exposure time, and increases to above 9.5 × 104 when exposure time increases to 7 h. This might be attributed to lightly cross-linked reaction of PMMA chain under UV light exposure for longer time. Based on the amount of PMMA brush layers determined by TGA and Mn derived from GPC, the graft density of PMMA can be estimated, and these results are summarized in Table 1.
 .
.Formation of hollow polymer vesicles
After removing the core of silica particle templates in mixture of acetone and HF aqueous solution, polymer vesicles can be obtained, whose morphology were revealed by TEM and SEM (Fig. 1g, h and 2e, f). TEM images indicate that at a high organic layer/silica ratio (SiO2@PDEAEMA@PMMA-7h), the hollow polymeric particles are robust and maintain their spherical shape. However, at low organic layer/silica ratios (SiO2@PDEAEMA@PMMA-3h), the resulting polymer shell is thin and flexible, and the hollow particles tend to deform. Asher and coworkers reported similar results when they synthesized hollow polymer particles of different core sizes.9 When the relatively thinner vesicles (PDMAEMA@PMMA-3h) were taken out of the solution and observed under extreme vacuum conditions in TEM and SEM, they tend to be deformed. However, the shapes of PDMAEMA@PMMA-7h keep basically intact and spherical, which might be ascribed to the thicker PMMA brush. This indicated that PMMA brush on the surface of polymer vesicles can protect polymer vesicles at some cases. For further confirmation of having fully removed the silica cores, FT-IR characterization was performed on etched hollow PDMAEMA@PMMA. The spectrum of PDMAEMA@PMMA shows no apparent Si–O bands at 1100 cm−1, verifying the complete removal of silica cores.Motivated by the well-defined structure, we tested the encapsulation properties of PDMAEMA@PMMA polymeric vesicles. As a common hydrophilic dye, methyl orange is insoluble in some organic solvents such as toluene. We added 3 mg of methyl orange and 3 mg vesicles in toluene and compared with a toluene solution only containing 3 mg of methyl orange. Encapsulation of dyes such as methyl orange with polymer vesicles could result in better stability in organic solvents. As shown in Fig. 5, the dye encapsulated by polymer visibly displayed better dispersion. The methyl orange precipitated from the stirred solution within 3 min. However, the dye encapsulated was stable, lasting for more than one day. It shows a demonstrable advance in encapsulation of dye, which leads to good dispersion in insoluble solvents. In addition, better colloidal stability is also worthy of remark because the polymer brush on the vesicles contributes to the prolonged and good dispersion.
 | ||
| Fig. 5 Photos of (a) 3 mg methyl orange and (b) 3 mg methyl orange/3 mg PDMAEMA@PMMA-7h, in 5 ml toluene. | ||
Conclusion
In summary, by using silica particles as templates, we demonstrated a facile and general approach for fabricating polymer vesicles with well-defined polymer brush through surface-initiated photopolymerization. This robust approach is expected to be very potential in fabrication and modification of polymer vesicles with different sizes and functions.Acknowledgements
The authors thank the National Nature Science Foundation of China (No: 50803036) and Science & Technology Commission of Shanghai Municipal Government (No: 08520 704700) for their financial support.References
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- I. Gill and A. Ballesteros, J. Am. Chem. Soc., 1998, 120, 8587–8598 CrossRef CAS.
- L. Zhang, R. Gao, M. Yang, X. Q. Jiang and B. Liu, Adv. Mater., 2007, 19, 2988–2992 CrossRef CAS.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- S. F. Chong, A. Sexton, R. D. Rose, S. J. Kent, A. N. Zelikin and F. Caruso, Biomaterials, 2009, 30, 5178–5186 CrossRef CAS.
- A. M. Yu, I. Gentle, G. Q. Lu and F. Caruso, Chem. Commun., 2006, 2150–2152 RSC.
- C. Boyer, M. Whittaker, C. Nouvel and T. Davis, Macromolecules, 2010, 43, 1792–1799 CrossRef CAS.
- G. L. Li, G. Liu, E. T. Kang, K. G. Neohnag and X. L. Yang, Langmuir, 2008, 24, 9050–9055 CrossRef CAS.
- X. L. Xu and S. A. Asher, J. Am. Chem. Soc., 2004, 126, 7940–7945 CrossRef CAS.
- T. Morinaga, M. Ohkura, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 2007, 40, 1159–1164 CrossRef CAS.
- M. S. Wong, J. N. Cha, K. S. Choi, T. J. Deming and G. D. Stucky, Nano Lett., 2002, 2, 583–587 CrossRef CAS.
- A. Koide, A. Kishimura, K. Osada, W. D. Jang, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2006, 128, 5988–5989 CrossRef CAS.
- A. S. Utada, E. Lorenceau, D. R. Link, P. D. Kaplan and D. A. Weitz, Science, 2005, 308, 537–541 CrossRef.
- M. Kobašlija and D. T. McQuade, Macromolecules, 2006, 39, 6371–6375 CrossRef CAS.
- A. Musyanovych and K. Landfester, Prog. Colloid Polym. Sci., 2008, 134, 120–127 CAS.
- D. W. Xiao, Z. X. Shen, T. Sand, X. B. Cheng, M. F. Li, L. Y. Chen and Z. S. Wang, J. Colloid Interface Sci., 2010, 341, 23–29 CrossRef CAS.
- C. S. Peyratout and L. Dähne, Angew. Chem., Int. Ed., 2004, 43, 3762–3783 CrossRef.
- L. A. Connal, C. R. Kinnane, A. N. Zelikin and F. Caruso, Chem. Mater., 2009, 21, 576–578 CrossRef CAS.
- T. K. Mandal, M. S. Fleming and D. R. Walt, Chem. Mater., 2000, 12, 3481–3487 CrossRef CAS.
- A. M. Rouhi, Chem. Eng. News, 1999, 77, 51–59.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS.
- D. E. Kataoka and S. M. Trolan, Nature, 1999, 402, 794–797 CrossRef CAS.
- A. Hucknall, S. Rangarajan and A. Chilkoti, Adv. Mater., 2009, 21, 2441–2446 CrossRef CAS.
- R. C. Bailey and J. T. Hupp, Anal. Chem., 2003, 75, 2392–2398 CrossRef CAS.
- M. Kaholek, W. K. Lee, J. Feng, B. D. LaMattina, J. Dyer and S. Zauscher, Chem. Mater., 2006, 18, 3660–3664 CrossRef CAS.
- C. Cheng, E. Khoshdel and K. L. Wooley, Nano Lett., 2006, 6, 1741–1746 CrossRef CAS.
- M. D. Rowe, B. A. G. Hammer and S. G. Boyes, Macromolecules, 2008, 41, 4147–4157 CrossRef CAS.
- R. Schmidt, T. Zhao, J. B. Green and D. J. Dyer, Langmuir, 2002, 18, 1281–1287 CrossRef CAS.
- X. Y. Jia, X. S. Jiang, R. Liu and J. Yin, ACS Appl. Mater. Interfaces, 2010, 2, 1200–1205 CrossRef CAS.
- F. T. Chen, X. S. Jiang, R. Liu and J. Yin, ACS Appl. Mater. Interfaces, 2010, 2, 1031–1037 CrossRef CAS.
- Y. N. Wen, X. S. Jiang, R. Liu and J. Yin, Polymer, 2009, 50, 3917–3923 CrossRef CAS.
- W. Stöber and A. J. Fink, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- X. S. Jiang and J. Yin, Chem. Commun., 2005, 4927–4928 RSC.
- X. S. Jiang and J. Yin, Macromolecules, 2004, 37, 7850–7853 CrossRef CAS.
- M. W. Louie, H. W. Liu, M. H. C. Lam, T. C. Lau and K. W. Lok, Organometallics, 2009, 28, 4297–4307 CrossRef CAS.
- J. X. Feng, R. T. Haash and D. J. Dyer, Macromolecules, 2004, 37, 9525–9537 CrossRef CAS.
- D. J. Dyer, J. X. Feng, R. Schmidt, V. N. Wong, T. F. Zhao and Y. Yagci, Macromolecules, 2004, 37, 7072–7074 CrossRef CAS.
- M. Ejaz, Y. Tsujii and T. Fukuda, Polymer, 2001, 48, 6811–6815 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |