A study on the hydrogen bonding interaction of the electrospun ladder polyphenylsilsesquioxane/polyisophthalamide composite fibers by ATR FT-IR†
Zhongjie
Ren
a,
Rongben
Zhang
b,
Feng
Wang
*a and
Shouke
Yan
*a
aState Key laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing, China. E-mail: skyan@mail.buct.edu.cn; Fax: +861064455928; Tel: +861064455928
bLaboratory of Polymer Science and Material, Beijing National Laboratory for Molecular Sciences (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing, China
First published on 9th November 2010
Abstract
Hydrogen bonds formed in ladder polyphenylsilsesquioxane (Ph-LPSQ)/polyisophthalamide (Cn-PA) composites and their electrospun fibers were studied by ATR FT-IR in detail. It has been found that the peak position of hydrogen bonded N–H stretching vibration in the electrospun composite fibers shifts to higher wavenumber compared to that produced in the composite powders. Moreover, the wavenumber shift increases with increasing electrospinning voltage. This is caused by the transformation of N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds into N–H⋯OSi2 ones during the electrospinning process. Possible structure models for the composite powders and the electrospun composite fibers were proposed.
C hydrogen bonds into N–H⋯OSi2 ones during the electrospinning process. Possible structure models for the composite powders and the electrospun composite fibers were proposed.
Introduction
Hydrogen bonding represents the dominant intermolecular interaction in a large number of natural and synthetic polymers. It could influence the glass transition temperature, melting temperature and crystallization behaviours of the polymers, and therefore, is essential to understanding the equilibrium structure and properties of these macromolecules.1,2 Many of the known three-dimensional chain structures of synthetic polymers and biological macromolecules exhibit highly ordered structures stabilized by hydrogen bonding involving intermolecular, intramolecular, or both interactions. For example, the well-known β-sheet structures of linear aliphatic polyamides (nylons) are based on the intermolecular N–H⋯OC hydrogen bonding between amide groups in adjacent chains. In addition, it is well known that hydrogen bonding is very easily affected by the ambient environment conditions such as temperature, solvent and so on. A variety of studies about the temperature and solvent effects on hydrogen bonding have been reported.3,4
It is noteworthy that the electrospinning technique can produce continuous polymer nanofibers from polymer solution or melt in high electric fields.5–7 Thus far, a wide variety of organic polymer nanofibers have been successfully prepared via this process.8,9 During the electrospinning process, under the application of an electric field, a drop of polymer solution is presented at the spinneret tip. As the intensity of the electric field is increased, mutual charge repulsion on the drop surface increases, which alters the droplet shape dramatically to form a Taylor cone.10 Eventually, charge repulsion exceeds surface tension and a jet of solution is ejected from the Taylor cone towards the grounded target substrate. During jet acceleration towards the substrate, substantial solvent evaporation leaves behind polymer fibers in the form of non-woven mats. Generally, the jet undergoes a whipping process during acceleration, which stretches the fiber and significantly reduces fiber diameter.11 During electrospinning the hydrogen bonding may be influenced. However, to the best of our knowledge, there is no detailed research about the effect of electrospinning on hydrogen bonding.
IR spectroscopy is sensitive to polymer microstructure, and has been widely used in investigating hydrogen bonding in polymer materials. Actually, a few transmission IR spectroscopy studies of polyphenylsilsesquioxane (Ph-LPSQ)12,13 and polyisophthalamide (Cn-PA)14,15 have already been reported. In the case of fibers, attenuated total reflectance (ATR) FT-IR is ideally suited as a tool to obtain structure information,16–18 since clear transmission spectra are difficult to collect due to light scattering and filament surface reflection. In this study, the hydrogen bonds formed in Ph-LPSQ/Cn-PA composites and their electrospun nanofibers are investigated in detail by ATR FT-IR.
Results and discussion
The formation of intermolecular hydrogen bonding between Si–O–Si bonds of Ph-LPSQ and N–H bonds of Cn-PA is expected in the composites of Ph-LPSQ/Cn-PA since the formation ability of hydrogen bonding between the Si–O–Si and N–H bonds has once been confirmed for the POSS/polypeptide system.19 It was reported that the N–H stretching vibration peak is very sensitive to hydrogen bonding and contains much useful structural information.20 Therefore, we are primarily concerned with the N–H stretching vibration mode. Fig. 1 presents the ATR FT-IR spectra of the C12-PA and its 50/50 Ph-LPSQ/C12-PA blend in the range 3100–3600 cm−1. From Fig. 1, we see that the C12-PA shows an absorption peak at 3269 cm−1, which is assigned to the hydrogen-bonded N–H stretching vibration peak, indicating the formation of hydrogen bond between the C12-PA. When blending the Ph-LPSQ with the C12-PA, this absorption peak shifts from 3269 cm−1 to 3285 cm−1 (see spectrum b in Fig. 1). At the same time, a broadening of the N–H stretching vibration band is observed in the Ph-LPSQ/C12-PA. The peak width at half-height measured for the Ph-LPSQ/C12-PA is somewhat larger than that of the C12-PA. According to the literature,20 the N–H stretching vibration is considered to be an isolated mode and normal coordinate calculation indicates that the potential energy distribution is essentially composed solely of this stretching motion. Accordingly, it is not a conformationally sensitive mode. Instead, the broadness of the hydrogen-bonded N–H band reflects, in large part, a distribution of hydrogen-bonded N–H groups of various strengths dictated by distance and geometry. Therefore, the above result indicates the formation of hydrogen bonding between the Si–O–Si bonds of Ph-LPSQ and N–H bonds of C12-PA. As a consequence, the Ph-LPSQ/C12-PA composite contains different kinds of hydrogen bondings, i.e. the intermolecular and intramolecular hydrogen bondings between carbonyl and amine groups of PA and the hydrogen bondings between Si–O–Si and N–H bonds. This increases the distribution of the hydrogen-bonded N–H groups with various strengths.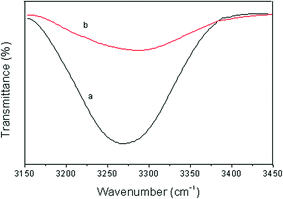 | ||
| Fig. 1 ATR FT-IR spectra of (a) C12-PA and (b) Ph-LPSQ/C12-PA (50/50) blend in the range 3100–3600 cm−1. | ||
To study the effect of electrospinning on the hydrogen bonding behavior of the Ph-LPSQ/Cn-PA, Ph-LPSQ/Cn-PA fibers have been prepared by electrospinning under optimal conditions. As an example, Fig. 2 illustrates the electrospun Ph-LPSQ/C12-PA fibers collected from a 23 wt% chloroform solution under different electrospinning voltages. It was found that beaded fiber structure was obtained when the electrospinning voltage was set at 8 kV. With elevated electrospinning voltage, uniform fibers with smooth surface have been produced. The average diameters of the fibers were found to be varied from 1.2 μm at 10 kV to 0.45 μm at 20 kV.
 | ||
| Fig. 2 SEM micrographs of Ph-LPSQ/C12-PA electrospun fibers from 23% chloroform solution with different electrospinning voltages. (A) 20 kV, (B) 15 kV, (C) 10 kV, and (D) 8 kV. | ||
The ATR FT-IR spectra of the Ph-LPSQ/Cn-PA composite fibers and powder are compared in the range 2600–3600 cm−1. As presented in Fig. 3, the peaks corresponding to the CH2 symmetric and asymmetric stretching mode at 2850 and 2960 cm−1 show the same band position and shape for both fiber and powder samples. This demonstrates that CH2 symmetric and asymmetric stretching mode is unaffected by the electrospinning. On the contrary, the N–H stretching band in the Ph-LPSQ/Cn-PA powder, as summarized in Table 1, varies from 3253 cm−1 in Ph-LPSQ/C6-PA to 3291 cm−1 in Ph-LPSQ/C18-PA. It illustrates a significant high wavenumber shift of the N–H absorption following the increase in the length of the substituted alkoxyl groups of PA. This indicates that bulky side groups of the PA inhibit the formation of hydrogen bondings in the composite by limiting the ability of the two chains to arrange and orient themselves correctly for forming stable hydrogen bondings.21,22
 | ||
| Fig. 3 ATR FT-IR spectra in the range of 2600–3600 cm−1 of Ph-LPSQ/C6-PA powder (a) and fiber (b); Ph-LPSQ/C12-PA powder (c) and fiber (d); and Ph-LPSQ/C18-PA powder (e) and fiber (f). | ||
| Sample | ν N–H/cm−1 | Aν N–H/AνCH2 |
|---|---|---|
| Ph-LPSQ/C6-PA powder | 3253 | 0.64 |
| Ph-LPSQ/C6-PA fiber | 3278 | 0.61 |
| Ph-LPSQ/C12-PA powder | 3289 | 0.48 |
| Ph-LPSQ/C12-PA fiber | 3318 | 0.46 |
| Ph-LPSQ/C18-PA powder | 3291 | 0.39 |
| Ph-LPSQ/C18-PA fiber | 3319 | 0.41 |
The band corresponding to the N–H stretching mode in the Ph-LPSQ/Cn-PA fibers also shifts to a higher wavenumber with increase in the length of substituted alkoxyl groups, which reflects the same effect of the side alkoxyl groups. However, a higher wavenumber shift of the N–H stretching absorption in the Ph-LPSQ/Cn-PA fiber as compared to the powder sample is recognized. This leads to a change in the N–H stretching absorption of ca. 25–29 cm−1. Crystallization may affect the hydrogen-bonded N–H stretching mode. It is, however, excluded since both the Ph-LPSQ/Cn-PA powder and its corresponding electrospun fiber are amorphous as confirmed by XRD (Fig. S1†). Another explanation rests on the molecular chains stretched by high acceleration voltage during the electrospinning, which may cause the dissociation or scission of the N–H hydrogen bond to give more free (non-hydrogen-bonded) N–H groups, and therefore result in N–H stretching mode shift to the higher wavenumber. To verify this possibility, the integrated area of the peak assigned to the N–H stretching vibration should be compared. Considering that the absorption coefficient and the sample thickness are different for the different composites, a sole comparison of the peak area of N–H stretching mode is meaningless. Therefore, the ratio of the integrated area of the N–H stretching peak (AνN–H) with that of the CH2 stretching peak (AνCH2) was conducted and compared under the consideration that the CH2 stretching peak is not affected by electrospinning as mentioned above. As can be seen from Table 1, for Ph-LPSQ/Cn-PA powder samples, the ratio of AνN–H and AνCH2 gradually decreases with the increase in length of the substituted alkoxyl group of PA. The ratio varies from 0.64 of Ph-LPSQ/C6-PA to 0.39 of Ph-LPSQ/C18-PA, indicating that the peak area of νN–H decreases with the increase of alkoxyl chain length. The ratio remains, however, almost the same for each Ph-LPSQ/Cn-PA powder and fiber samples with the same Cn-PA, suggesting the relative total peak area of νN–H is unaffected by electrospinning. Therefore, to clarify the effect of electrospinning on the hydrogen bond, the subsequent proportional calculation of area for free N–H groups in total N–H groups should be considered.
From above description of the experimental results, it does appear that there are three district contributions to the N–H absorption region, i.e. the absorptions of free N–H groups and the two kinds of hydrogen bonded N–H groups. To check the change in free N–H groups before and after electrospinning, we only resort to using peak and curve fitting. We have severely restricted the options by making a number of justifiable assumptions to obtain the best fit. First, the frequency of the band attributed to the hydrogen bonded and free N–H groups are readily established from a second-derivative plot of the obtained spectra. Second, the band shape was assumed to be Gaussian. Third, a linear baseline was assumed from 3100 to 3600 cm−1. And the curve fitting was limited to the spectral data available between 3100 and 3600 cm−1.
The representative curve fitting results of the Ph-LPSQ/C12-PA samples are shown in Fig. 4. For powder and fiber samples, only two bands are necessary to obtain a satisfactory fitting of the experimental data. This is very close to the results obtained previously for the amorphous nylon.23 The acquired two bands are at approximately 3320 and 3440 cm−1 assigned to the hydrogen-bonded and free N–H stretching vibration respectively,24 which is pleasingly consistent with the second-derivative data (see Fig. 4b and d). However, the acquired two bands vary with the Ph-LPSQ/Cn-PA and show obvious higher wavenumber shifts with the longer alkoxyl groups. As shown in Table 2, for the composite powders the acquired two bands increases from 3320 and 3440 cm−1 in Ph-LPSQ/C6-PA to 3325 cm−1 and 3445 cm−1 in Ph-LPSQ/C18-PA. The same increasing tendency is observed for the composite fibers.
 | ||
| Fig. 4 Curve-fitting results of N–H stretching vibration of Ph-LPSQ/C12-PA. (a) Ph-LPSQ/C12-PA powder; (b) second-derivative spectrum of (a); (c) Ph-LPSQ/C12-PA fiber; (d) second-derivative spectrum of (c). | ||
| Sample | A 3440 (%)a | ν N–H, associated/cm−1b | ν N–H, free/cm−1c |
|---|---|---|---|
| a Peak area of free N–H stretch. b Peak position of hydrogen bonded N–H stretch. c Peak position of free N–H stretch. Note: A3320 = 100% − A3440. | |||
| Ph-LPSQ/C6-PA powder | 18.2 | 3320 | 3440 |
| Ph-LPSQ/C6-PA fiber | 19.0 | 3323 | 3443 |
| Ph-LPSQ/C12-PA powder | 22.5 | 3324 | 3441 |
| Ph-LPSQ/C12-PA fiber | 22.1 | 3327 | 3444 |
| Ph-LPSQ/C18-PA powder | 26.1 | 3325 | 3445 |
| Ph-LPSQ/C18-PA fiber | 25.3 | 3328 | 3447 |
As summarized in Table 2, for Ph-LPSQ/Cn-PA powder, the peak area at ca. 3440 cm−1 increases from 18.2% for Ph-LPSQ/C6-PA to 26.1% for Ph-LPSQ/C18-PA, which confirms that the large alkoxyl groups prevent the formation of hydrogen bonding. However, the peak areas of the hydrogen-bonded N–H and free N–H groups in Ph-LPSQ/Cn-PA are almost unchanged before and after electrospinning. Therefore, the band shift of the hydrogen-bonded N–H band reflects only a different distribution of the hydrogen-bonded N–H groups of varying strengths. As mentioned above, there are two kinds of hydrogen bonding in the Ph-LPSQ/Cn-PA system: N–H⋯OC and N–H⋯OSi2. The hydrogen bond between Si–O–Si and N–H bonds is weaker than that between CO and N–H bonds. So the former makes the shift of wavenumber higher compared with the latter.25,26
It occurred to us that the hydrogen bonding between the CO and N–H bonds reduces while the hydrogen bonding between the Si–O–Si and N–H bonds increases during the electrospinning process. A possible mechanism is shown in Scheme 1. The Ph-LPSQ is a double chain rigid polymer, which should be extended before and after electrospinning. But the Cn-PA chains can fold because the alkoxyl groups increase the flexibility of molecular chains. The state of the Ph-LPSQ/Cn-PA composite powder is described in Scheme 1a. As mentioned in the literature,27 the composites were prepared in situ and the molar ratio of the Ph-LPSQ and Cn-PA is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. So the Ph-LPSQ and Cn-PA chains may distribute uniformly. In the powder, the Cn-PA chains is random coil (Scheme 1a), which promotes the formation of hydrogen bonding of N–H⋯OC. However, Cn-PA chains may be stretched under high voltage during electrospinning, which results in the Cn-PA chains transformed into relatively unwinded state from the random coil. In this situation, the probability of forming the N–H⋯OSi2hydrogen bonding is increased with respect to the N–H⋯OC hydrogen bonding, as shown in Scheme 1b. Formation of more N–H⋯OSi2hydrogen bonds made the peak position of the N–H stretching mode shift to higher wavenumber.
:1. So the Ph-LPSQ and Cn-PA chains may distribute uniformly. In the powder, the Cn-PA chains is random coil (Scheme 1a), which promotes the formation of hydrogen bonding of N–H⋯OC. However, Cn-PA chains may be stretched under high voltage during electrospinning, which results in the Cn-PA chains transformed into relatively unwinded state from the random coil. In this situation, the probability of forming the N–H⋯OSi2hydrogen bonding is increased with respect to the N–H⋯OC hydrogen bonding, as shown in Scheme 1b. Formation of more N–H⋯OSi2hydrogen bonds made the peak position of the N–H stretching mode shift to higher wavenumber.
 | ||
| Scheme 1 A proposed model about hydrogen bond of Ph-LPSQ/Cn-PA. (a) Before electrospinning and (b) after electrospinning. | ||
Moreover, since the fiber morphology depends strongly on the electric field voltage used during electrospinning, ATR FT-IR spectra of Ph-LPSQ/C12-PA fibers prepared under four different conditions were recorded as shown in Fig. 5. The regions of the Si–O–Si stretching mode and N–H stretching mode are focused. Similarly, the peak position and shape of the CH2 symmetric and asymmetric stretching mode at 2850 and 2960 cm−1 are unchanged when the electronic field voltage increases. Not surprisingly, the peak maximum of the N–H stretching mode increases with the electronic field voltage and shifts from 3289 cm−1 of 0 kV (Fig. 5a) over 3310 cm−1 of 10 kV (Fig. 5c) to 3318 cm−1 of 20 kV (Fig. 5e). The region in the range of 1000–1200 cm−1 assigned to the Si–O–Si stretching mode has also notable change with the increase of the voltage. According to the literature,28,29 the stretching mode of Si–O–Si of ladder polysilsesquioxanes displays two absorption bands around 1050 and 1150 cm−1 associated with symmetric vibration (νSi–O–Si(ring-sym)) and asymmetric stretching vibrations (νSi–O–Si(ring-asym)) of (Si–O)4 subunits, respectively. In our studied Ph-LPSQ/C12-PA composite, two stretching absorption of the Si–O–Si in Ph-LPSQ located at around 1047 and 1135 cm−1 was also found as shown in Table 3 and Fig. 5. The band at around 1047 cm−1 assigned to symmetric vibration of (Si–O)4 subunits is very sensitive to the microstructure28 and its shape and peak maximum change obviously with the increase of electronic field voltage. But the band at 1135 cm−1 is nearly invariable. Interestingly, the band of symmetric vibration of (Si–O)4 subunits of Ph-LPSQ/C12-PA at 1047 cm−1 shows a shoulder peak at around 1030 cm−1, which should be caused by the formation of hydrogen bonding between N–H and Si–O–Si groups. Moreover, the relative intensity of the peak at around at 1047 cm−1 decreases gradually with the increase of the electrospinning voltage and shifts from 1047 cm−1 of 0 kV to 1041 cm−1 of 20 kV. On the contrary, the relative intensity of the peak at 1030 cm−1 increases with the electrospinning voltage, suggesting the relative content of the N–H⋯OSi2hydrogen bonding increases.
 | ||
| Fig. 5 ATR FT-IR spectra of Ph-LPSQ/C12-PA fiber in the range 2600–3600 and 1000–1200cm−1 with different electrospinning voltage (a) 0 kV; (b) 8kV; (c) 10 kV; (d) 15 kV; (e) 20 kV. | ||
| Voltage/kV | ν Si–O– Si(ring-sym) /cm−1 | ν Si–O– Si(ring-asym) /cm−1 | ν N–H/cm−1 |
|---|---|---|---|
| 0 | 1047, 1030 | 1135 | 3289 |
| 8 | 1046, 1030 | 1135 | 3305 |
| 10 | 1044, 1030 | 1135 | 3312 |
| 15 | 1043, 1030 | 1135 | 3316 |
| 20 | 1041, 1030 | 1135 | 3318 |
Pick peak and curve fitting of (Si–O)4 subunits symmetric vibration region and N–H stretching region of Ph-LPSQ/C12-PA with different electrospinning voltages also are carried out. The same justifiable assumptions are made as mentioned above. The representative curve fitting results of (Si–O)4 subunits symmetric vibration region for Ph-LPSQ/C12-PA are shown in Fig. 6 and Table 4. For different electrospinning voltages samples, two bands are still obtained to satisfy fitting of the experimental data. The acquired two bands are at approximately 1030 and 1045 cm−1 respectively, which is also consistent with the second-derivative data (see Fig. 6c,d). As shown in Table 4, when the electrospinning voltages increase, the acquired two bands decreases from 1031 and 1047 cm−1 of 0 kV to 1027 cm−1 and 1041 cm−1 of 20 kV. Moreover, the peak area at ca. 1030 cm−1 increases from 35.3% of 0 kV to 53.2% of 20 kV for Ph-LPSQ/C12-PA. According to the discussion above, the peak at around 1030 cm−1 assigne to N–H bonded (Si–O)4 subunits symmetric vibration, so this result shows the relative content of the N–H…OSi2hydrogen bonding increases.
 | ||
| Fig. 6 Curve-fitting results of (Si–O)4 subunits symmetric vibration region of Ph-LPSQ/C12-PA with different electrospinning voltages. (a) 10 kV; (b) 20 kV; (c) second-derivative spectrum of (a); (d) second-derivative spectrum of (b). | ||
In addition, The curve fitting results for the N–H stretching region of Ph-LPSQ/C12-PA with different electrospinning voltages are shown in Table 5. On one hand, the peak area and positions at 3440 cm−1 attributed to free N–H stretching are almost constant with the increase of the electrospinning voltages, suggesting the free N–H groups are unaffected by the electrospinning voltage. On the other hand, the peak area at ca. 3320 cm−1 is also unchanged with the increasing electrospinning voltages. But the peak position shifts to the higher wavenumber. A shift from 3324 cm−1 of 0 kV to 3330 cm−1 of 20 kV shows the N–H⋯OSi2hydrogen bonding increases with the electrospinning voltage. This result confirmed the proposed mechanism again.
Conclusions
The hydrogen bonding developed in ladder polyphenylsilsesquioxane (Ph-LPSQ)/polyisophthalamide (Cn-PA) composites and their electrospun fibers were studied in detail by ATR FT-IR. The peak position of the hydrogen bonded N–H stretching vibration in electrospun fibers shifts to higher wavenumber than that in composite powders. Moreover, the wavenumber shift increases with increasing electrospinning voltage. However, the free N–H groups are unaffected by the electrospinning as estimated through curve fitting. The transformation of hydrogen bonding from N–H⋯OC into N–H⋯OSi2 happens during the electrospinning process. A possible mechanism was proposed.
Experimental
The composites of polyphenylsilsesequioxane/poly(5-alkoxylisophthalamide) (Ph-LPSQ/Cn-PA), with Cn–PA being the poly(5-hexyloxyisophthalamide) (C6-PA), poly(5-dodecyloxyisophthalamide) (C12-PA) and poly(5-octadecyloxyisophthalamide) (C18-PA), were prepared according to our published paper.27DSC measurements demonstrate that the Cn-PA in the fibers is in the amorphous state.The electrospun Ph-LPSQ/Cn-PA composite fibers on the aluminium foils were used for the ATR-FTIR measurement. For comparison, the Ph-LPSQ/Cn-PA composite powder was dissolved in THF and then solution cast onto the aluminium foils for the ATR-FTIR measurement.
Infrared spectra were acquired on a Perkin Elemer SP100 Fourier transform infrared spectrometer with a PIKE Technologies Horizontal ATR attachment (KRS-5 internal reflection crystal having an incident angle of 45°). All of the spectra were recorded in the range of 400–4000 cm−1 at a resolution of 4 cm−1 and were baseline corrected using spectrometer software. A minimum of 64 scans were signal averaged.
The electrospinning apparatus consisted of a metered flow pump, a high D.C. voltage supply and aluminium foils as targets for fiber collection. Electrospinning solutions were prepared by dissolving the required polymers on weight percentage basis in the solvent and stirring the solutions for 24 h to make a well mixed homogenous solution. The solution was then ejected through a syringe using a syringe flow pump at feed rate of 1 ml h−1 within different electric field with constant tip target distance of 20 cm.
Acknowledgements
The financial support of NSFC (No. 50521302, 50833006, 50973008 and 50773088) and Open Project of State Key Laboratory of Supramolecular Structure and Materials (SKLSSM 201016) is gratefully acknowledged.Notes and references
- W. O. Baker and C. S. Fuller, J. Am. Chem. Soc., 1942, 64, 2399–2407 CrossRef CAS.
- D. Garcia and H. W. Starkweather, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 537–555 CrossRef CAS.
- M. M. Coleman, L. A. Narvett and P. C. Painter, Polymer, 1998, 39, 5867–5869 CrossRef CAS.
- Y. He, N. Asakawa and Y. Inoue, Macromol. Chem. Phys., 2001, 202, 1035–1043 CrossRef CAS.
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS.
- Y. Dzenis, Science, 2004, 304, 1917–1919 CrossRef CAS.
- Y. Zhu, J. Zhang, Y. Zheng, Z. Huang, L. Feng and L. Jiang, Adv. Funct. Mater., 2006, 16, 568–574 CrossRef CAS.
- S. Kedem, J. Schmidt, Y. Paz and Y. Cohen, Langmuir, 2005, 21, 5600–5604 CrossRef CAS.
- L. Li, S. Limmer, S. Seraji, Y. Tammy, P. Chou, C. Nguyen and G. Cao, Adv. Funct. Mater., 2002, 12, 59–64 CrossRef CAS.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1964, 280, 383–397 CrossRef.
- Y. M. Shin, M. M. Hohman, M. P. Brenner and G. C. Rutledge, Appl. Phys. Lett., 2001, 78, 1149–1151 CrossRef CAS.
- J. F. J. R. Brown, L. H. Jr. Vogt and P. I. Prescott, J. Am. Chem. Soc., 1964, 86, 1120 CrossRef CAS.
- Q. L. Zhou, S. K. Yan, P. Xie and R. B. Zhang, Acta. Polym. Sin., 2007, 10, 918–930 Search PubMed.
- E. G. Marta, M. J. Mosquera, M. A. Amelia and M. D. T. Juan, Chem. Mater., 1994, 6, 1918–1924 CrossRef.
- P. K. Kim, S. L. Hsu and H. Ishida, Macromolecules, 1985, 18, 1905–1914 CrossRef CAS.
- V. Premnath, Macromolecules, 1995, 28, 5139–5143 CrossRef CAS.
- C. S. P. Sung, Macromolecules, 1981, 14, 591–594 CrossRef CAS.
- J. P. Hobbs and C. S. P. Sung, Macromolecules, 1983, 16, 193–199 CrossRef CAS.
- S. W. Kuo, L. H. Fang, W. J. Huang, K. U. Jeong and F. C. Chang, Macromolecules, 2009, 42, 1619–1626 CrossRef CAS.
- D. J. Skrovanek, S. E. Howe, P. C. Painter and M. M. Coleman, Macromolecules, 1985, 18, 1676–1683 CrossRef CAS.
- M. M. Coleman, G. J. Pehlert and P. C. Painter, Macromolecules, 1996, 29, 6820–6831 CrossRef CAS.
- D. J. Skrovanek, P. C. Painter and M. M. Coleman, Macromolecules, 1986, 19, 699–705 CrossRef CAS.
- L. R. Schroeder and S. L. Cooper, J. Appl. Phys., 1976, 47, 4310–4315 CrossRef CAS.
- A. Okada, M. Kawasumi, I. Tajima, T. Kurauchi and O. Kamigaito, J. Appl. Polym. Sci., 1989, 37, 1363–1371 CrossRef CAS.
- M. F. Roberts and S. A. Jenekhe, Macromolecules, 1991, 24, 3142–3146 CrossRef CAS.
- K. H. Lee, K. W. Kim, A. Pesapane, H. Y. Kim and J. F. Rabolt, Macromolecules, 2008, 41, 1494–1498 CrossRef CAS.
- Z. Ren, Y. Tian, P. Xie, S. Yan and R. Zhang, Polym. Chem., 2010, 1, 1095–1101 RSC.
- E. S. Park, H. W. Ro, C. V. Nguyen, R. L. Jaffe and D. Y. Yoon, Chem. Mater., 2008, 20, 1548–1554 CrossRef CAS.
- H. Adachi, E. Adachi, O. Hayashi and K. Okahashi, Rep. Prog. Polym. Phys. Jpn., 1985, 28, 261–267 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD and ATR FT-IR data for the composites. See DOI: 10.1039/c0py00274g |
| This journal is © The Royal Society of Chemistry 2011 |