Monitoring Atom Transfer Radical Polymerisation using 14C-radiolabelled initiators†
Mark
Long
a,
Suzanne H.
Rogers
a,
David W.
Thornthwaite
a,
Francis R.
Livens
b and
Steve P.
Rannard
*c
aUnilever Research and Development Port Sunlight Laboratories, Quarry Road East, Bebington, Wirral, UK CH63 3JW
bSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, UK M13 9PL
cDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: srannard@liv.ac.uk; Fax: +44 (0)151 794 3501; Tel: +44 (0)151 794 3501
First published on 16th October 2010
Abstract
Radio thin layer chromatography (R-TLC) and liquid scintillation counting, of fractions collected from conventional gel permeation chromatography (GPC), have been used to study the fate of 14C-labelled initiators in the ambient methanolic ATRP of 2-hydroxypropyl methacrylate at different targeted number average degrees of polymerisation. Benzyl 2-bromoisobutyrate was synthesised in unlabelled form and with 14C-labels at different locations to establish no adverse effects of the radiolabel. Target chain lengths of 10, 25 and 50 monomer units were synthesised and comparison of GPC and R-TLC showed a significant under-utilisation of the initiator with approximately 16% clearly observable at high monomer conversion (>97%). New chains appeared to be initiated at monomer conversions >90% and as late as 300 minutes after polymerisation had commenced. Purification by repeated precipitation was shown to be superior to flash chromatography for the ability to remove residual unreacted or terminated initiator although increased fractionation could be seen with each repeat.
Introduction
Controlled polymerisations offer many advantages to the experimental polymer chemist including the determination of polymer chain length, low polydispersity, end group chemistry and polymer architecture including block copolymer, star polymer and graft polymer synthesis.1 To the polymer end-user, the accurate description and purity of polymer properties, including the chain length and chemical nature of the macromolecule, are important for structure–property relationships2 and descriptors for model generation.3The last thirty years have seen the introduction of a range of controlled syntheses including Group-Transfer Polymerisation (GTP),4 Reversible Addition–Fragmentation chain Transfer (RAFT) polymerisation,5Nitroxide-Mediated Polymerisation (NMP),6 “click” chemistry,7dendrimer synthesis,8 immortal polymerisation9 and Atom Transfer Radical Polymerisation (ATRP).10
Arguably, one of the most successful of these controlled polymerisation techniques is ATRP with reports of successful synthesis of branched,11 block,12 and star polymers,13polymerisation in emulsion conditions,14polymerisation using ionic liquids,15 hydrophobic solvents and aqueous environments,16 heterogeneous polymerisation from surfaces17 and modification of natural polymers,18 all using a range of monomer types. Many studies of ATRP mechanism have been reported19 but our recent report of utilising 14C-labelled initiators20 for ATRP remains the only direct radiochemical study of the fate of initiator during polymerisation.
Radiolabels have been utilised for many years to aid polymer detection in studies such as mucoadhesion21 or pharmaceutical delivery. Often polymer-labelling is achieved by a treatment of pre-formed polymers, for example tritiation of polymer protons,22 chelation of heavy metal radioisotopes23 (e.g.64Cu), methylation of amines24 using 14C-methyl iodide or iodination25 using 125I. Each of these treatments is, by necessity, a chemical reaction that has the potential to degrade the polymer or, at least, to modify polymer properties and behaviour. In comparison, substitution of 14C for existing carbons26 within the polymer building blocks (monomers/initiators) is probably the least intrusive labelling strategy. Selective labelling of polymer end-groups also avoids statistical label incorporation along the polymer chain, i.e. all labelled chains carry a single radioactive site. Often, the radiolabelling of polymers is conducted to allow detection of very low concentrations of material in complex environments27 (e.g. body fluids). The activity of the polymer allows a clear signal to be measured without interference from other chemical species and without the need for lengthy purification procedures that may alter the study sample. Herein, we describe our utilisation of 14C-radiolabelled ATRP initiators and demonstrate the insights achieved by accurate detection and monitoring of both polymer and residual initiator during purification and analysis.
Experimental
Reagents and suppliers
All reagents and solvents were purchased from the Sigma Aldrich Group unless otherwise stated. Prosafe Liquid Scintillation Cocktail was purchased from Meridian Biotechnologies; PMMA GPC Standards were purchased from Polymer Laboratories; Partisil LK6DF Silica Gel 60A TLC Plates were purchased from BDH; 14C methyl iodide (98.4%) was purchased from Amersham Biosciences; 14CH2 benzyl alcohol (95%) was kindly supplied by Unilever.Material synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu(I)Br:bipy ratio of 1:1:2 was used throughout. Identical polymerisations for each initiator were performed in triplicate to confirm reproducibility, demonstrate consistent polymerisation across labelled and non-labelled initiators and allow accurate radioactivity measurements. Kinetic experiments involved removal of 150 µl of the reaction mixture and aeration to quench the polymerisation at regular intervals prior to analysis without additional purification.
:Cu(I)Br:bipy ratio of 1:1:2 was used throughout. Identical polymerisations for each initiator were performed in triplicate to confirm reproducibility, demonstrate consistent polymerisation across labelled and non-labelled initiators and allow accurate radioactivity measurements. Kinetic experiments involved removal of 150 µl of the reaction mixture and aeration to quench the polymerisation at regular intervals prior to analysis without additional purification.
Silica flash chromatography . After 24 hours polymerisation, two 150 µl samples of the reaction mixture were removed for analysis without purification. 150 µl aliquots of the remaining reaction mixture were dried to remove methanol solvent and unreacted monomer, redissolved in THF and passed through a Kieselgel Merck Type 10180 Mesh 70–230 silica column.
Precipitation . 500 µl of the reaction mixture were added to dry methanol (2 ml). The mixture was then slowly added to deionized water (30 ml) whereupon the polymer formed a white precipitate. After sedimentation of the polymer, samples of the supernatant were removed for analysis prior to separation of the precipitate and two further repetitions of the process
Gel permeation chromatography, GPC. GPC analysis was conducted using an Agilent 11100 series refractive index detector, 400C Eppendorf oven, Jasco PU 1580 pump, Jasco AS 590 auto sampler, Polymer Laboratories PLgel 5 µm mixed-C and PLgel 5 µm mixed-D columns with a PL guard column. THF was used as the elution solvent at a flow rate of 0.8 ml min−1. Fractions of eluted solvent were collected over 1 minute intervals to enable radioactivity levels to be determined using liquid scintillation counting.
Liquid scintillation counting . Liquid scintillation counting was conducted using a Packard Tri Carb 3100 TRC liquid scintillation counter and utilised a 20 ml scintillation vial containing a known concentration of radioactive compound and 10 ml of Prosafe Liquid Scintillation Cocktail containing 60–75% phenyl xylyl ethane, 20–40% alcohol ethoxylate, 2–8% alcohol ether phosphate ester, 0.1–1.0% 2,5-diphenyloxazole and 0.1–1.0% 1,4-bis(4-methyl-α-styryl)benzene. Results are presented as Disintegrations Per Minute (DPM).
Radio thin layer chromatography (R-TLC). R-TLC analysis was conducted using an AR 2000 BIOSCAN Radio TLC imaging scanner utilising a gas filled proportional counter filled with 90/10 argon/methane to detect the β emissions from thin layer chromatography (TLC) plates. TLC samples (1.0% w/w) were eluted using Whatman TLC plates (Partisil LK6DF silica Gel 60A with indicator), with a silica thickness of 250 µm and 3 cm pre-adsorbent zone, to a standard distance.
Nuclear magnetic resonance (NMR) spectroscopy. 1H NMR and 13C NMR spectroscopy was conducted using a Brüker Avance DRX500 spectrometer. 1H NMR utilised approximately 2 mg of each sample dissolved in 1 ml of CDCl3; 13C NMR utilised approximately 50 mg of sample dissolved in 1 ml of CDCl3.
Results and discussion
When choosing to radiolabel a polymervia an ATRP initiator, two strategies are immediately obvious; the reaction of 2-bromoisobutyric acid, 1, with a 14C-labelled alcohol, Scheme 1A, or the synthesis of 14C-labelled 2-bromoisobutyric acid, 8, for esterification with hydroxyl-containing compounds, Scheme 1C.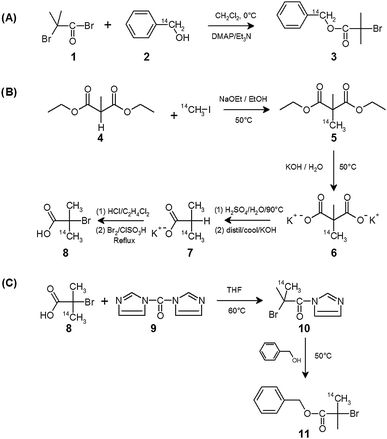 | ||
| Scheme 1 Synthesis of 14C-labelled ATRP initiators: (A) esterification of 14C-labelled benzyl alcohol, (B) synthesis of 14C-labelled 2-bromoisobutyric acid, (C) reaction of 14C-labelled 2-bromoisobutyric acid with benzyl alcohol. | ||
Radiolabelled materials are inherently energetic and undergo radioactive degradationviaradiolysis. Although the 14C isotope has a half-life of 5730 ± 40 years, β-decay results in conversion of 14C atoms to 14N atoms.26d As such, the formation of complementary initiators labelled in different positions allows confirmation of results and confidence in the integrity of the radiolabel during the timescale of the experiment. Utilisation of analogous non-labelled initiators allows comparison of polymerisations in the presence and absence of radiolabel to ensure no adverse impact of the radiolabel on the course of the polymerisation.
We have previously described our approach to the production of 14C-labelled initiators,203 and 11, via the reaction of 2-bromoisobutyric acid, 1, with 14C-labelled benzyl alcohol, 2, and, secondly, the synthesis of 14C-labelled 2-bromoisobutyric acid, 8, and subsequent reaction with benzyl alcohol, as summarised in Scheme 1.
These initiators were shown to initiate the ATRP of 2-hydroxypropyl methacrylate (2-HPMA) to produce near identical polymers. Gel permeation chromatography (GPC) and 1H nuclear magnetic resonance (NMR) spectroscopy of polymers with three target number average degrees of polymerisation (DPn) showed high conversion (>95%) with similar kinetics. The polydispersity (PDI) of each polymer was higher than expected, possibly due to the restricted solubility of the initiator in the methanol solvent chosen for the polymerisation but the values are within the reported range for ATRP conducted in protic solvents. Comparison with non-labelled initiator analogues, Table 1, showed near identical behaviour hence confirming the lack of impact on the ATRP mechanism of the radiolabels.
Radio-analysis of Atom Transfer Radical Polymerisation
Experimentally, polymerisationviaATRP can be considered as comprising three key steps: (1) initiator choice and synthesis, (2) polymer synthesis and (3) polymer purification. The presence of radiolabels allows a novel examination of these steps through focussing purely on the radiolabelled material within the complex mixtures that are present. Step 1 of initiator synthesis has been described above (Scheme 1) and will be discussed again later.During this study we chose to place the 14C-label at either the methyl group of the tertiary carbon, 11, or the benzylic methylene group, 3, Scheme 1, to allow for comparative studies and validation of results through the elimination of false data from initiator fragmentation. The synthesis of the initiator and the comparison of polymerisation with unlabelled initiators have been described previously,20 however, the radiolabel allows additional monitoring of the progress of the polymerisation through the radioanalysis of the samples taken for kinetic studies.
Each sample utilised for kinetic analysis was conventionally analysed using the GPC refractive index (RI) detector to establish number average molecular weight (Mn), weight average molecular weight (Mw) and PDI. Additionally, fractions were collected during GPC analysis from the eluted solvent, in one minute time portions, and measured using liquid scintillation counting. As such, the total radioactivity for each 60 second time slice can be determined for the eluted solvent. The resolution of such a measurement is restricted and is clearly not as accurate as the almost constant sampling by the RI detector, however, this analysis allows the detection and direct correlation of the radioactivity of the sample with the polymer concentration detection by RI. Fig. 1 shows the combined scintillation/RI GPC analysis of the 14C CH2-labelled initiator, 3. An amount of broadening has occurred during transit through the GPC column although the peak is sharp and corresponds well with the radioactivity trace. 60 second sampling was adopted to ensure appreciable levels of detectable radioactivity in each time slice. The resolution of such a measurement could be enhanced by increasing the specific activity of the radio-initiator, allowing higher activity within smaller volumes of eluted solvent.
 | ||
| Fig. 1 GPC analysis of benzyl 2-bromoisobutyrate (initiator 3) studied using refractive index detection (green trace; concentration detection) and scintillation counting of fractions from the GPC eluent (red bars; radioactivity detection). | ||
When polymer samples were analysed using this combined approach, the initiator radioactivity was overlayed with both the radioactivity of the polymer sample and the RI trace from the GPC. After polymerising for just 30 minutes at ambient temperature, using initiator 3, with a target DPn = 50 monomer units, a sample was taken and injected directly into the GPC with subsequent fractions taken from the eluent, Fig. 2A. The GPCRI chromatogram shows a distinct polymer peak with a retention time of ca. 19 minutes. Additional features include a peak at ca. 23.5 minutes, corresponding to unreacted monomer, and a clear peak at ca. 22 minutes. The monomer peak and the additional peak at 22 minutes were also clearly visible in the 30 minute sample taken from the polymerisation using initiator 11, with a 14C CH3-label. Radioactivity measurement of the eluting solvent shows a strong signal corresponding to the main polymer RI peak and a second peak in the radioactivity corresponding to the RI chromatogram signal at 22 minutes. This also corresponds directly with the radioactivity trace derived from the unreacted initiator 3, Fig. 1, showing a significant concentration of material that appears to be unreacted or terminated initiator.
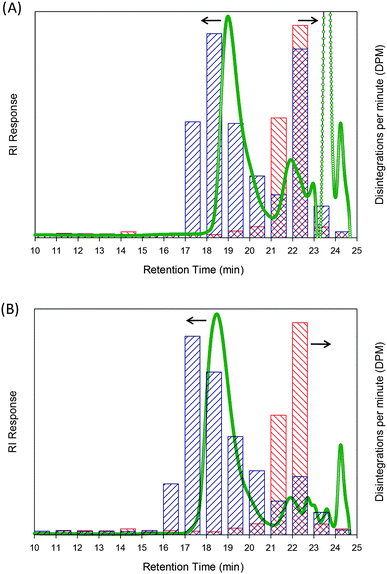 | ||
| Fig. 2 GPC analysis of ATRP of 2-HPMA using initiator 3. (A) Sample taken after 30 minutes polymerisation; (B) sample after 24 hours polymerisation. Refractive index (green trace; concentration detection) and scintillation counting of fractions collected from the GPC eluent (blue bars; radioactivity detection) are shown, overlayed with scintillation of fractions for initiator 3 (red bars) for comparison. | ||
Throughout the polymerisation the radioactivity of the initiator signal decreases progressively relative to polymer as new chains are formed. Fig. 2B shows the final polymer sample which had been left to polymerise for 24 hours. The peak corresponding to unreacted monomer (23.5 minutes) also reduced considerably, confirmed by our 1H NMR study of conversion (97.2%). However, although both the radioactivity and RI initiator signals corresponding to initiator have reduced, they did not fully disappear during the reaction, suggesting either that a portion of the initiator added at the outset of the polymerisation did not initiate chains or that the initiation led to rapid termination before appreciable propagation.
Radio thin-layer chromatography
Radio thin-layer chromatography (R-TLC) is an excellent tool for determining the radio-purity of labelled materials without separating non-labelled impurities.28 A conventional TLC experiment is conducted but the TLC plate is imaged using a proportional counter that is scanned along the plate, detecting radioactivity levels. As such, unlabelled materials are invisible to the scan and only the radiolabelled materials are detected after appropriate/conventional separation with solvent or solvent mixtures. Comparison to conventionally imaged unlabelled TLC under identical conditions confirms the compound assignment by reference to retardation factor (Rf) values.20 The R-TLC of 14CH3-labelled 2-bromoisobutyric acid is shown in Fig. 3A.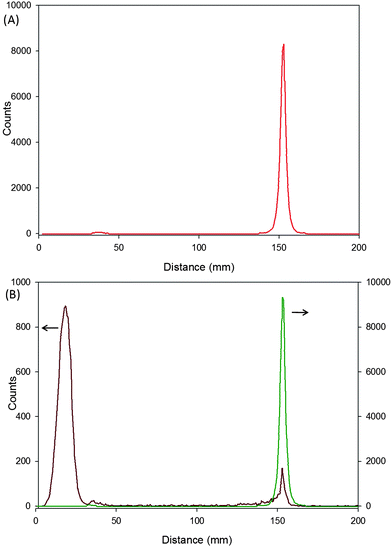 | ||
| Fig. 3 Radio thin layer chromatogram of: (A) 14C methyl-labelled 2-bromoisobutyrate (initiator 11; red trace); (B) 14C methylene-labelled 2-bromoisobutyrate (initiator 3; green trace) overlayed with poly(2-HPMA) synthesised from 3 with a target DPn = 50 monomer units (dark red trace), after 24 hours polymerisation time (>97% conversion); (eluent: Et2O–CH3CO2H 90/10 v/v.). | ||
The ability to monitor the radio-species without purification of the sample allows direct measurement without concern that sample purification has removed a significant amount of the material under investigation. The polymer samples taken for GPC kinetic evaluation and fraction collection/scintillation counting were also subject to R-TLC. Fig. 3B shows the R-TLC of the unpurified polymer after polymerisation for 24 hours and an overlayed R-TLC of the 14C CH2-labelled initiator 3 for comparison. It is clear that the initiator peak has been adequately separated from the polymer under these conditions.
As previously described, the activity of the signal from the unreacted/terminated initiator can be compared to the total activity of the R-TLC and the proportion of initiator not leading to significant propagation can be measured directly. The R-TLC measurements of unreacted/terminated initiator can be directly compared to the radioactivity observed within the GPC fraction analysis. R-TLC leads to a clear separation of the initiator from the polymer although the GPC columns used during this study were not able to separate completely the low molecular weight tail of the polymer from the initiator and the resolution of the fractions did not adequately allow a baseline activity to be determined between the polymer and initiator. Nonetheless, by selecting regions of elution defined by the fraction collection of the initiator (GPC) or the detected peak definition (R-TLC), it was possible to estimate the amount of unreacted/terminated initiator throughout the polymerisation indicated by each method.
Fig. 4 shows a comparison of polymerisations using both initiator 3, Fig. 4A, and 11, Fig. 4B, with a target DPn = 50 monomer units. Both polymerisations reach very similar conversions (>95% by 1H NMR) and PDIs (approximately Mw/Mn = 1.4). Interestingly, both reactions consume monomer and initiator at similar rates but neither utilises all of the available initiator; indeed after 30 minutes, approximately 30% of the initiator appears to be unreacted or terminated. Although the GPC appears to estimate a higher amount of unused initiator in both polymerisations, probably reflecting the limited resolution of GPC fraction collection and inadequate separation, the trends follow the R-TLC results with good correlation.
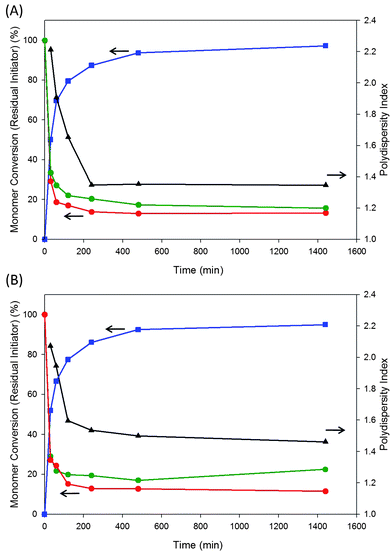 | ||
| Fig. 4 Comparison of R-TLC (red trace), GPC/liquid scintillation (green trace), 1H NMR (conversion; blue trace) and GPC/RI (PDI; black trace) analysis of ambient ATRP of 2-HPMA (target DPn = 50) with benzyl 2-bromoisobutyrate in methanol. (A) Methylene labelled initiator 3 and (B) methyl labelled initiator 11. | ||
When averaged across the polymerisations and the analytical techniques, the percentage of initiator not utilised in polymer chain formation during the polymerisation is approximately 16%, consistent with our earlier report. Surprisingly, it is also evident from this analysis that initiator continues to be consumed at least up to 200 minutes after the polymerisation has nominally commenced. There is also evidence to suggest that new chains are being initiated after 300 minutes of polymerisation and monomer conversions of >90%.
The results shown here for the monitoring of the ambient methanolic ATRP of 2-HPMA initiated by benzyl 2-bromoisobutyrate are clearly specific to this monomer within this particular solvent and using this initiator. The analysis does, however, suggest that, within an ATRP, the accurate targeting of DPn, with low PDI at high monomer conversion, through control of the ratio of monomer and initiator is not without complication, as seen in many reports. The poor utilisation of initiator will inevitably lead to the formation of polymers with higher Mn than targeted but the continual initiation of new chains in the latter stages of monomer conversion will also lead to broad PDIs. It has become common to report PDIs > 1.2 as “near monodisperse” within ‘controlled radical’ polymerisations but such values would indicate considerable loss of polymerisation control in classical ‘living’ polymerisations such as anionic polymerisation.
The radiolabelled material that we have quantified by R-TLC and GPC fractionation/liquid scintillation counting has been cautiously assigned as unreacted or terminated initiator as we have no chemical analysis of the structure of the compounds involved. The R-TLC shows Rf values that correspond directly to the initiators 3 and 11, Fig. 3 and 5, and terminated initiator should have considerably different Rf values determined by the different polarities and solubilities derived from their different chemical structures. Combined with the GPC/scintillation counting it is compelling evidence that the signals derive directly from unreacted initiator but at this stage we cannot be definitive that this is the case.
 | ||
| Fig. 5 Radio thin layer chromatograms of: (A) poly(2-HPMA) synthesised from 3 with a target DPn = 50 monomer units (initiator shown as green trace for comparison): polymer sample at 24 hours polymerisation time (>97% monomer conversion) (dark red trace), polymer sample after silica flash chromatography (red trace), polymer sample after three precipitations (blue trace); (B) overlayed supernatant of repeated precipitations: precipitation supernatant 1 (red trace), precipitation supernatant 2 (blue trace), precipitation supernatant 3 (green trace)—R-TLC data have been normalised to trace initiator peak to show comparative peak height with removed polymer. (Eluent: Et2O–CH3CO2H 90/10 v/v.) | ||
Radio-analysis of polymer purification
As discussed earlier, a key stage in the synthesis of polymers is purification to remove unreacted monomer, initiator and catalyst residues. This is especially critical in ATRP as organometallic catalysts are often employed. Very commonly, copper is a contaminant that is readily observed in ATRP samples as the polymers may be slightly green or blue in colour. Residual copper and monomer may lead to issues of toxicity if the often complex block copolymers are designed for use in biomedical applications and several techniques to minimise or completely remove metal-based catalysts have been reported.29 Sufficient but incomplete removal of metal catalyst residues is often achieved by passing an ATRP polymer through a column of silica or alumina. The metal is retained on the column and ideally a clear/colourless solution yielding a pure white polymer sample after solvent removal. Alternatively polymers may be precipitated into non-solvents to remove monomer residues and metal contaminants. It would also be expected that other contaminants such as unreacted initiator or terminated oligomers would also be removed during these procedures.The polymers produced with radiolabelled initiators were subjected to both flash chromatography through silica columns and subsequent repeated precipitation from methanol solution into deionised water. R-TLC was used at each stage to study the ability of each step to remove unreacted/terminated initiator. As can be seen from Fig. 5A, flash chromatography led to a limited increase in the radiopurity of the polymer with respect to unreacted/terminated initiator within the target DPn = 50 polymer samples. Quantification of the activity attributable to unreacted/terminated initiator is shown in Table 2.
| Initial initiator radioactivity (average, %) | Initiator radioactivity post-silica column (average, %) | Initial DPn (1H NMR calc. monomer units) | DPn post-silica column (1H NMR calc. monomer units) | |
|---|---|---|---|---|
| Poly(2-HPMA) (target DPn = 50) | 7.67 | 6.85 | 37 | 55 |
| Poly(2-HPMA) (target DPn = 25) | 11.83 | 13.18 | 30 | 26 |
| Poly(2-HPMA) (target DPn = 10) | 36.12 | 36.78 | 8 | 7 |
The R-TLC suggests that reliance on simple flash chromatography for purification may provide benefits for catalyst removal but non-polymeric species related to initiator are not significantly removed and therefore will contribute to inaccuracies in DPn confirmation by 1H NMR end-group analysis. Indeed, the lower chain length samples exhibited a small but noticeable increase in radioactivity from the initiator impurity after flash chromatography, probably due to a higher removal of oligomeric species by the column relative to the initiator removal at these lower chain lengths, Table 2.
Characterisation of the polymers by 1H NMR after passage through the silica column was compared with data obtained prior to flash chromatography, Table 2. The removal of initiator residues that was observed through R-TLC of the DPn = 50 polymer sample indeed led to a considerable increase in the calculated DPn to approximately 55 monomer units. The observed relative increase in initiator residue for both the polymers with target DPn = 25 and 10 monomer units led to a subsequent proportional decrease in calculated DPn by end-group analysis, presumably through the weighted removal of oligomers relative to initiator and the resulting discrepancy through the inability to discriminate between end-groups and initiator contaminants using 1H NMR.
Precipitation of the polymer samples after flash chromatography led to a distinct removal of non-polymeric radiolabelled material and the reduction of initiator residues, Fig. 5A, but full removal was not seen even after three precipitations, Table 3.
| Initiator radioactivity post-silica column (average, %) | Initiator radioactivity post-precipitation (average, %) | DPn post-silica column (1H NMR calc. monomer units) | DPn post-precipitation (1H NMR calc. monomer units) | |
|---|---|---|---|---|
| Poly(2-HPMA) (target DPn = 50) | 6.85 | 3.5 | 55 | 72 |
| Poly(2-HPMA) (target DPn = 25) | 13.18 | 4.47 | 26 | 39 |
| Poly(2-HPMA) (target DPn = 10) | 36.78 | 31.53 | 7 | 14 |
Increased fractionation of the polymer sample was clearly observed as the R-TLC of the supernatant after each precipitation showed an increase in the presence of polymer relative to removed initiator. In combination with initiator removal, fractionation would be expected to have a profound effect on the determination of DPn by 1H NMR spectroscopy as the removal of both oligomers and unreacted/terminated initiator leads to an assessment of higher average polymer chain lengths. Table 3 shows the effect of precipitation on each of the different target DPn polymers within our study.
Conclusions
14C-Radiolabelling of initiators allows the controlled introduction of radioactivity into polymer molecules and the monitoring of initiator fate during the course of a polymerisation without the need to purify or extract the initiator from the reaction mixture. Within the ATRP investigation detailed here, we have shown that the benzyl 2-isobutyrate ATRP initiator (3 and 11) continues to be consumed throughout the first 200–300 minutes of the polymerisation of 2-HPMA in methanol at ambient temperature. The initiator is not fully utilised leading to a residue identifiable by radiometric methods within both the R-TLC and the GPC eluent fractions. Progressive and slow initiator consumption throughout the polymerisation leads to the continued initiation of new chains and suggests a plausible explanation for the difficulty in achieving very low PDIs at high monomer conversion and for the often reported attainment of higher DPn than targeted using ATRP. Although similar slow consumption of ATRP initiators has been reported for polyethylene glycol (PEG) macroinitiators,30 the previous studies showed complete reaction of each initiator and related initiator efficiency to increasing steric hinderance derived from increased PEG chain length.Purification techniques such as silica column flash chromatography and polymer precipitation have variable impact on the final sample, dependent on the DPn of the synthesised polymer. Flash chromatography does little to remove residual initiator but may impact the lower molecular weight fractions of the polymer distribution, generating misleading molecular weight information through NMR end-group analysis. Precipitation is an excellent technique for initiator residue removal but the expected fractionation of low molecular weight species impacts the final recovered molecular weight distribution.
Further work is required to study different ATRP initiator/monomer/solvent combinations to establish the general nature of our findings. The strategies for radiolabelling ATRP polymers presented here have application in the monitoring and study of other new polymerisation approaches such as RAFT and NMP. The final radiolabelled materials also allow the detection of polymers in complex environments and further work will endeavour to establish new insights of the behaviour of well defined polymers in a range of applications.
Acknowledgements
The authors wish to thank the Royal Society for an Industry Fellowship (SR) and Unilever for financial support (ML). Megan E. M. Rannard is also warmly thanked for help compiling graphical data.References
- (a) M. Szwarc, ACS Symp. Ser., 1981, 166, 1 CAS; (b) K. Matyjaszewski, ACS Symp. Ser., 2009, 1023, 3 CAS; (c) J. Jagur-Grodzinski, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2116 CrossRef CAS; (d) A. Hirao, S. Loykulnant and T. Ishizone, Prog. Polym. Sci., 2002, 27, 1399 CrossRef CAS; (e) R. Godoy Lopez, F. D'Agosto and C. Boisson, Prog. Polym. Sci., 2007, 32, 419 CrossRef; (f) H. S. Bisht and A. K. Chatterjee, J. Macromol. Sci., Polym. Rev., 2001, 41, 139.
- (a) K.-I. Izutsu and K. Shigeo, Phys. Chem. Chem. Phys., 2000, 2, 123 RSC; (b) N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS; (c) S. Jana, S. P. Rannard and A. I. Cooper, Chem. Commun., 2007, 2962 RSC.
- For example: D. N. Theodorou, Mol. Phys., 2004, 102, 147 Search PubMed.
- (a) O. W. Webster, Adv. Polym. Sci., 2004, 167, 1 CAS; (b) O. W. Webster, W. R. Hertler, D. Y. Sogah, W. B. Farnham and T. V. RajanBabu, J. Am. Chem. Soc., 1983, 105, 5706 CrossRef CAS.
- (a) R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad and S. H. Thang, Macromolecules, 1999, 32, 6977 CrossRef CAS; (b) C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402 CrossRef CAS; (c) S. L. Brown, C. M. Rayner, S. Graham, A. Cooper, S. Rannard and S. Perrier, Chem. Commun., 2007, 2145 RSC.
- (a) B. Charleux, ACS Symp. Ser., 2003, 854, 438 CAS; (b) N. R. Cameron, C. A. Bacon and A. J. Reid, ACS Symp. Ser., 2003, 854, 452 CAS.
- (a) B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 15020 CrossRef CAS; (b) M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS.
- (a) D. A. Tomalia and J. M. J. Frechet, Prog. Polym. Sci., 2005, 30, 217 CrossRef CAS; (b) F. Aulenta, W. Hayes and S. Rannard, Eur. Polym. J., 2003, 39, 1741 CrossRef CAS; (c) W. J. Feast, S. P. Rannard and A. Stoddart, Macromolecules, 2003, 36, 9704 CrossRef CAS.
- (a) S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861 CrossRef CAS; (b) M.-L. Hsueh, B.-H. Huang and C.-C. Lin, Macromolecules, 2002, 35, 5763 CrossRef CAS.
- (a) T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC; (b) S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS; (c) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS.
- (a) T. He, D. J. Adams, M. F. Butler, C. T. Yeoh, A. I. Cooper and S. P. Rannard, Angew. Chem., Int. Ed., 2007, 46, 9243 CrossRef CAS; (b) T. He, D. J. Adams, M. F. Butler, A. I. Cooper and S. P. Rannard, J. Am. Chem. Soc., 2009, 131, 1495 CrossRef CAS; (c) I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483 CrossRef CAS.
- (a) J. V. M. Weaver, Y. Tang, S. Liu, P. D. Iddon, R. Grigg, N. C. Billingham, S. P. Armes, R. Hunter and S. P. Rannard, Angew. Chem., Int. Ed., 2004, 43, 1389 CrossRef CAS; (b) E. S. Read, K. L. Thompson and S. P. Armes, Polym. Chem., 2010, 1, 221 RSC.
- (a) P. T. Hammond, Macromolecules, 2009, 42, 368 CrossRef; (b) H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960 CrossRef CAS.
- (a) K. Min, H. Gao, J. A. Yoon, W. Wu, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2009, 42, 1597 CrossRef CAS; (b) G. Chambard, P. De Man and B. Klumperman, Macromol. Symp., 2000, 150, 45 CrossRef CAS.
- (a) M. Xie, Y. Kong, H. Han, J. Shi, L. Ding, C. Song and Y. Zhang, React. Funct. Polym., 2008, 68, 1601 CrossRef CAS; (b) T. Biedron and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2799 CrossRef CAS.
- (a) X.-S. Wang, S. F. Lascelles, R. A. Jackson and S. P. Armes, Chem. Commun., 1999, 1817 RSC; (b) S. McDonald and S. P. Rannard, Macromolecules, 2001, 34, 8600 CrossRef CAS.
- (a) C. Perruchot, M. A. Khan, A. Kamitsi, S. P. Armes, T. von Werne and T. E. Patten, Langmuir, 2001, 17, 4479 CrossRef CAS; (b) C.-D. Vo, A. Schmid, S. P. Armes, K. Sakai and S. Biggs, Langmuir, 2007, 23, 408 CrossRef CAS.
- (a) J. Lindqvist and E. Malmström, J. Appl. Polym. Sci., 2006, 100, 4155 CrossRef CAS; (b) D. Bontempo, G. Masci, P. De Leonardis, L. Mannina, D. Capitani and V. Crescenzi, Biomacromolecules, 2006, 7, 2154 CrossRef CAS; (c) S. P. Rannard, S. H. Rogers and R. Hunter, Chem. Commun., 2007, 362 RSC.
- (a) T. Pintauer and K. Matyjaszewski, Top. Organomet. Chem., 2009, 26, 221 CAS; (b) P. A. Gurr, M. F. Mills, G. G. Qiao and D. H. Solomon, Polymer, 2005, 46, 2097 CrossRef CAS; (c) F. Stoffelbach, R. Poli, S. Maria and P. Richard, J. Organomet. Chem., 2007, 692, 3133 CrossRef CAS.
- M. Long, D. W. Thornthwaite, S. H. Rogers, G. Bonzi, F. R. Livens and S. P. Rannard, Chem. Commun., 2009, 6406 RSC.
- M. E. A. McGirr, S. M. McAllister, E. E. Peters, A. W. Vickers, A. F. Parr and A. W. Basit, Eur. J. Pharm. Sci., 2009, 36, 386 CrossRef CAS.
- J. C. Russell, J. R. Jones, T. A. Vick and P. W. Stratford, J. Labelled Compd. Radiopharm., 1993, 33, 957 CAS; C. Postolache, L. Matei and R. Georgescu, J. Radioanal. Nucl. Chem., 2009, 280, 251 CrossRef CAS.
- G. Sun, J. Xu, A. Hagooly, R. Rossin, Z. Li, D. A. Moore, C. J. Hawker, M. J. Welch and K. L. Wooley, Adv. Mater., 2007, 19, 3157 CrossRef CAS.
- O. Oulanti, J. Widmaier, E. Pefferkorn, S. Champ and H. Auweter, J. Colloid Interface Sci., 2005, 291, 98 CrossRef CAS.
- (a) N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Frechet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842 CrossRef CAS; (b) E. R. Gillies, E. Dy, J. M. J. Frechet and F. C. Szoka, Mol. Pharmaceutics, 2005, 2, 129 CrossRef CAS.
- (a) R. K. Zeidan, S. A. Kalovidouris, T. Schluep, R. Fazio, R. Andresini and M. E. Davis, Bioconjugate Chem., 2006, 17, 1624 CrossRef CAS; (b) G. Shemilt, M. Newby and S. L. Kitson, J. Labelled Compd. Radiopharm., 2007, 50, 515 CrossRef CAS; (c) R. G. Riley, J. D. Smart, J. Tsibouklis, J. A. Davis, G. Kelly, F. Hampson, P. W. Dettmar and W. R. Wilber, J. Biomed. Mater. Res., 2001, 58, 102 CrossRef CAS; (d) J. N. Huffaker, Nucl. Phys. A, 1971, 171, 87 CrossRef CAS.
- (a) J. Kucka, M. Hruby and O. Lebeda, Appl. Radiat. Isot., 2010, 68, 1073 CrossRef CAS; (b) K. Saatchi and U. O. Hafeli, Bioconjugate Chem., 2009, 20, 1209 CrossRef CAS.
- E. Gattavecchia, D. Tonelli, A. Breccia, A. Fini and E. Ferri, J. Radioanal. Nucl. Chem., 1994, 181, 77 CAS.
- For example (a) L. Mueller and K. Matyjaszewski, Macromol. React. Eng., 2010, 4, 180 Search PubMed; (b) M. E. Honigfort, W. J. Brittain, T. Bosanac and C. S. Wilcox, Macromolecules, 2002, 35, 4849 CrossRef CAS; (c) Y. Shen, H. Tang and S. Ding, Prog. Polym. Sci., 2004, 29, 1053 CrossRef CAS.
- S. Perrier and D. M. Haddleton, Eur. Polym. J., 2004, 40, 2277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methodology, NMR spectra, mass spectra and R-TLC data. See DOI: 10.1039/c0py00275e |
| This journal is © The Royal Society of Chemistry 2011 |