Robust bonding and one-step facile synthesis of tough hydrogels with desirable shape by virtue of the double network structure†
Junji
Saito
a,
Hidemitsu
Furukawa
bc,
Takayuki
Kurokawa
bd,
Rikimaru
Kuwabara
a,
Shinya
Kuroda
a,
Jian
Hu
a,
Yoshimi
Tanaka
b,
Jian Ping
Gong
*b,
Nobuto
Kitamura
e and
Kazunori
Yasuda
e
aGraduate School of Science, Hokkaido University, Sapporo, 060-0810, Japan
bFaculty of Advanced Life Science, Hokkaido University, Sapporo, 060-0810, Japan. E-mail: gong@sci.hokudai.ac.jp; Fax: +81-11-706-2774; Tel: +81-11-706-2774
cDepartment of Mechanical Systems Engineering, Graduate School of Science and Engineering, Yamagata University, Yonezawa, 992-8510, Japan. E-mail: furukawa@yz.yamagata-u.ac.jp; Fax: +81-238-26-3197; Tel: +81-238-26-3197
dCreative Research Initiative Sousei, Hokkaido University, Sapporo, 001-0021, Japan
eDepartment of Sports Medicine and Joint Surgery, Hokkaido, University Graduate School of Medicine, Sapporo, 060-8638, Japan. E-mail: yasukaz@med.hokudai.ac.jp; Fax: +81-11-706-7822
First published on 9th November 2010
Abstract
Robust bonding of a hydrogel in aqueous environment, either to another hydrogel or to a solid, is one of the major unsolved issues for the practical applications of hydrogels in various fields. Here we report robust bonding between a pair of hydrogel sheets, containing over 90 wt% of water, by applying the double-network (DN) structure. In the optimal condition, the peeling energy of the united gel sheets reaches 1200 J m−2, which is comparable to the bulk fracture energy of a normal type of tough DN gels. This hydrogel bonding technique is also applied to form tough bonding between hydrogel and plastic plates. Furthermore, based on this technique, we have developed a facile method to synthesize robust double network hydrogels with any desirable free-shape from micro-gel precursors. These novel techniques will substantially merit the applications of the tough hydrogels in various fields, such as an artificial meniscus.
Introduction
Polymer hydrogels had been too brittle and too weak to be used as any load-bearing materials until the beginning of 2000. Since 2001, several different approaches to improve extraordinarily the mechanical strength of hydrogels had been proposed.1–7 Our group have developed novel high-strength and high toughness hydrogels3 by introducing a strongly asymmetric double network (DN) structure inside the hydrogels, which were composed of a tightly cross-linked polyelectrolyte gel that is highly swollen in water as the first network, and a sparsely cross-linked neutral polymer that is densely packed in the polyelectrolyte gel as the second network. This kind of asymmetric structure is obtained via a two-step sequential free-radical polymerization process of the two networks. The DN gels synthesized under an optimal condition, despite of over 90 wt% water content, exhibit extraordinary mechanical performances in terms of its high Young's modulus (∼0.3 MPa), fracture stress (∼20 MPa), and fracture energy (∼1000 J m−2),3,8–11 which correspond to the performances of the human articular cartilage. The inventions of these hydrogels with high mechanical performance substantially broaden their potential application in various fields, for example, as substitute of articular cartilage,12 scratchproof coating materials,13cell–sheet cultivation,14 antifouling materials,15etc.In the practical applications of hydrogel systems, some problems still remain: (1) weak bonding of a hydrogel in aqueous environment, either to another hydrogel or to a solid by using conventional glues due to the high water content of gels; (2) low freedom to form various desirable shapes. For example, due to the two-step sequential polymerization process of a DN gel, it is not possible to synthesize a tough DN gel with a complicated shape, such as the shape of a meniscus in human joint; (3) poor processability, especially for the DN gels. The first polyelectrolyte network that is highly swollen in water is usually too brittle and weak to handle. Furthermore, the two-step polymerization process in the preparation of the DN gels is very time consuming since the monomer diffusion of the second component is involved in the preparation. These problems hinder the practical application and large-scale production of DN gels. Here we report a breakthrough, which solves these problems simultaneously. The breakthrough starts from the invention of the strong bonding between swollen hydrogels, taking advantage of toughening mechanism of the DN gels. Based on this invention of bonding, we further develop a simple technique to prepare high-strength DN gels from particle gel precursors. These achievements, with no doubt, will substantially promote the industrial based large-scale production and various applications of high-strength gels, such as medical transplantation, tissue engineering, and lab-on-a-chip technology.
Experimental
Preparation of normal DN gels and bonded DN gels
2-Acrylamido-2-methylpropanesulfonic acid (AMPS or ATBS) was a courtesy from Toagosei Co., Ltd. and used as received. Acrylamide (AAm) (Junsei Chemical Co. Ltd) was recrystallized from chloroform. N,N′-Methylenebis (acrylamide) (MBAA; Tokyo Kasei Co., Ltd.), as a cross-linker for both AMPS and AAm gels, was recrystallized from ethanol. 2-Oxoglutaric acid (Wako Pure Chemical Industries, Ltd.), as an UV initiator for the gelation reactions, was used as received. Normal DN gels are synthesized through a two-step sequential free-radical polymerization.3 In the first step for the first network preparation, MBAA (4 mol%) and 2-oxoglutaric acid (0.1 mol%) were added to 1 M AMPS solution (1 M) (the molar percentages, 4 and 0.1 mol%, are determined by the ratio to the AMPS monomer). The solution was poured into reaction cells consisting of a pair of glass plates with a 2 mm spacing. Photopolymerization was carried out under argon atmosphere with an UV lamp for 10 h. In the second step for the second network preparation, after the gelation was completed, the PAMPS gels were immersed into 2 M AAm aqueous solution containing 2-oxoglutaric acid (0.01 mol%) and MBAA (0–0.25 mol%) for at least 1 d until the equilibrium was reached. Then photopolymerization was carried out for the PAMPS gels swollen in aqueous monomer solution with UV lamp for 10 h, and the normal DN gels were obtained. For the case of gel–gel bonding, a pair of PAMPS gel sheets, swollen in aqueous AAm monomer solution, were contacted to each other under a prescribed normal stress and irradiated with UV lamp for 10 h in the same way, and the adhered DN gel sheets were obtained.Preparation of particle-DN (P-DN) gel
PAMPS gels were prepared by photopolymerization in the same way as the normal DN gel preparation. As-prepared PAMPS gels were roughly grinded by spoon into particles and dried by using vacuum oven for 24 h. After this, the dried particles were minutely grind by mortar and pestle to become the particle size below several tens of micrometres. Then, the dried PAMPS particles were immersed into AAm aqueous solution (2 M) containing 2-oxoglutaric acid (0.01 mol%) and MBAA (0–0.25 mol%), where the volume ratio of AAm solution to PAMPS particles was made about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to form paste. Then the paste was poured into molds and photopolymerization was carried out with UV lamp for 8 h, and the P-DN gels were obtained.
:1 to form paste. Then the paste was poured into molds and photopolymerization was carried out with UV lamp for 8 h, and the P-DN gels were obtained.
Fracture energy and bonding energy measurement
The fracture energy of bulk gel was measured by a tear test described in the previous paper.9 DN gels were cut into the shape that is of standardized JIS-K6252 1/2 size (w = 5–5.5 mm, d = 7.5 mm, L = 30 mm, initial notch = 20 mm), with a gel cutting machine (Dumb Bell Co., Ltd). The two arms of a test piece were cramped and one of the arms was pulled at a constant velocity and the tearing force was recorded with a commercial test machine (Tensilon RTC-1150A, Orientec Co.). Fracture energy, G, defined as the energy required to create a unit area of fracture surface in a sample gel, is calculated by the following equation [note†], G = Fave/w, where Fave is the average tearing resistance force, and w is the width of the gel. All the samples were measured at a constant tearing velocity of 4.2 × 10−3 m s−1 (500 mm min−1) for the evaluation of both the peeling and fracture energies.Results and discussion
Fig. 1a shows strong bonding of two pieces of PAMPS gel sheets by polymerizing the second network PAAm inside both of the PAMPS gel sheets and their interface. At first, the two pieces of the first network PAMPS gel sheets are prepared. Then, the prepared PAMPS gel sheets were dipped in the aqueous solution of the AAm monomer of the second network gels. After the gel sheets reach their equilibrium state in the aqueous solution, the two gel sheets were contacted and the second network PAAm gel was polymerized by UV irradiation. Thus, a united DN gel is prepared. Fig. 1b schematically shows the structure of the united gels by forming a virtue of double network structure. | ||
| Fig. 1 Robust bonding between hydrogels. (a) Photograph of the united hydrogel sheets prepared by applying the double network (DN) structure. The lower gel sheet was colored by yellowish green pigment for the eye. (b) Schematic illustration of the formation of the DN structure between a pair of PAMPS gel sheets by synthesizing PAAm gel inside both the PAMPS gel sheets and the interface. The PAAm gel is also slightly cross-linked with the PAMPS gels by residual unsaturated vinyl bonds of the PAMPS gels. The dashed circles in the PAAm polymer indicate the characteristic expanding length of PAAm polymer chain. | ||
The strength of the united bonding strongly depends on the preparation conditions. Fig. 2 shows the effect of the cross-linker density in the second polymerization on the peeling energy which is defined as the energy to create a unit fractured surface between the two bonded gel sheets. When no or little amount (0.001 mol%) of the cross-linker was used, the peeling energy was as small as a bulk single network (∼6 J m−2).9 However, when the second cross-linker density increased to 0.01 mol%, the peeling energy steeply increased to a value as large as the fracture energy of the normal DN gels (∼600 J m−2). It should be noted that in this condition it is hard to recognize whether the fracture tip does run along the bonded surface or not. It implies that the peeling energy of the bonding interface of the united DN gels become comparable to the fracture energy of the bulk of the normal DN gels. After the peeling energy reached the maximum value, it decreased as the second cross-linker concentration increased, which is the similar to the decreasing tendency of the normal DN gels. As far as we know, such a strong bonding between hydrogels had not been achieved before. Under an optimized condition, the united interface is formed quite uniformly, and it is hardly recognized by the eye, because the gels keep their transparency under the second polymerization.
 | ||
| Fig. 2 Peeling energy of united DN gel in comparison with the fracture energy of the normal DN gels, as functions of the cross-linking density of the second network. The united DN gel was prepared with a negligible normal compression. | ||
Before starting discussions on the bonding of gels, we make a short review of the mechanism why the DN gels have such anomalously high toughness. Based on many previous studies on the DN gels,3,8,11,17–23 the high toughness of the DN gels during the large deformation is caused by the emergence of damaged zone around a crack tips. In the fracture process of the DN gels, the stress concentrating at the crack tip efficiently causes an internal fracture around the crack tip, which dissipates an anomalously large amount of fracture energy. The internal fracture of the DN gels comes from the breaking process of the brittle PAMPS network, while the ductile PAAm network chains stretch and keep entangled with the fractured PAMPS gel with no breaking until a quite large deformation. For the efficient breaking process of the PAMPS gels, the interaction between the PAMPS and the PAAm networks in the double-network structure is crucial, where the cross-linker plays an important role in such an interaction. Quite recently, we have elucidated that in the first PAMPS network gels a small amount of residual double bond (0.06 mol% in usual PAMPS gels) remains and reacts with AAm to form the chemical cross-linking between the first PAMPS network and the second PAAm network during the second polymerization.16 This is why the toughest DN gel is obtained when no additional cross-linker is added for the polymerization of the second PAAm network.
Why does the increase of the cross-linker concentration induce the steep decrease of the fracture energy? It has been revealed that an excess cross-linker suppresses the internal fracture that dissipates huge amount of fracture energy.17 It implies that the delicate balances among a suitable brittleness of the first PAMPS network, a suitable ductility of the second PAAm network, and a suitable interaction between the first and the second gels, should be preserved to achieve the anomalously high-strength by the DN structure.
Based on the above toughening mechanism of the DN structure, we readily know that the very low peeling strength of the bonded DN gel at low cross-linker density is related to a poor contact24,25 between the two PAMPS gel sheets. Here let us consider the contact of the two pieces of ideally flat PAMPS gels in water. According to electrostatic double layer (EDL) theory, an electrostatic double layer is formed at the interface of the two like-charged PAMPS gels. This EDL prevents the direct contact of the PAMPS gels. Therefore, it is difficult for the PAAm to form a continuously connected network at the interface in the case of low cross-linker density. In order to solve this problem, a compressive stress was applied on the contacting sheets of PAMPS during second polymerization. Fig. 3 shows the effect of normal stress on peeling energy of the united DN gel without adding any cross-linker in the second polymerization of PAAm. The peeling energy increases as the normal stress increases. It implies that the increase of the normal stress improves the contact between the surfaces.
 | ||
| Fig. 3 Peeling energy of the united DN gels as a function of d/h, where d is the equivalent random coil diameters of PAAm and h is the calculated electrostatic double layer thickness in pure water between two PAMPS gel sheets. Normal stress was changed as (i) 2 kPa, (ii) 30 kPa, and (iii) 140 kPa for Mw ≈ 5 × 105 g mol−1, (iv) 2 kPa for Mw ≈ 1 × 107 g mol−1, where Mw is the molecular weight of PAAm. The broken line is the guide for the eye. The PAAm network was synthesized without adding any additional cross-linker. | ||
If the PAAm molecules are much longer than the EDL, then the PAAm chain can be fixed to the PAMPS network through the residual double bond of PAMPS, and the bonding strength is also improved. For example, when we increase the average molecular weight from 8.8 × 105 g mol−1 to 3.4 × 107 g mol−1, which can be done by decreasing the initiator concentration in polymerizing AAm,26,27 the peeling energy increased steeply from 8 J m−2 to 100 J m−2.
Based on the our previous study, if the normal stress is at 2 kPa, 30 kPa, and 140 kPa, the electrostatic double layer thickness, i.e. the gel–gel distance between PAMPS gels, h is estimated as 66 nm, 17 nm, and 8 nm, respectively.28 On the other hand, the characteristic size of the second network chain can be also estimated as a function of the molecular weight. According to our previous work, the equivalent random coil diameters d of PAAm are calculated as 47 nm and 245 nm for the molecular weight of 5 × 106 g mol−1 and 1 × 107 g mol−1.26,29 Then, we plot the peeling energy as a function of the ratio of two characteristic lengths d/h. Fig. 3 obviously shows that the peeling energy linearly increases with d/h. It suggests that, in order to obtain the large peeling energy of the united DN gels, we should aim to increase the value of d/h.
These results simply imply that the increase of d/h increases the probability that the second PAAm chain forms multi-covalent bonds to the two PAMPS network sheets by chemically reacting with un-reacted residual cross-linkers of the PAMPS networks. Also, Fig. 2 shows that above about the 0.01 mol% cross-linker concentration, the peeling energy of the united DN gels became corresponding with the fracture energy of the usual robust DN gels. It implies that by using a suitable amount of cross-linker, the united structure develops to connect the two gel sheets robustly, regardless of the compressive pressure and molecular weight (d/h). Here it should be noted that an effective enhancement of the adhesion between hydrogels was previously achieved by a sensible way of polymer interdiffusion, i.e. by the interpenetration of linear polymer chain into both the hydrogels.30 Our approach is, however, conceptually advanced because the anomalous enhancement of the peeling energy is achieved by applying the DN-structure to the interface between hydrogels.
The DN-structure technique is also applied to form united and tough interface between hydrogel and plastic plates. Fig. 4 shows the DN gel–polyethylene substrate interface, bonded by introducing the DN structure. The polyethylene substrate has porous structure, of which pore size is several tens microns. Before bonding, the first PAMPS gels were prepared in the pore. All the pores of the substrate were filled with the first PAMPS gels. Then, the PAMPS gel sheet and the porous substrate were immersed in the aqueous solution of the AAm monomer of the second network PAAm gels. The gel sheet and the substrate were contacted and the second network PAAm gel was polymerized by UV irradiation. In this way, a robust gel–solid plate interface was developed and the peeling energy of interface became several hundred J m−2. A detailed study on the binding between DN gel and porous solids has been carried out recently.31
 | ||
| Fig. 4 Robust bonding of the DN hydrogel on a porous polyethylene plate, the plate is 3 mm in thickness and slightly bended by the swelling of the DN gel. | ||
Furthermore, as a result of these experiments about the united bonding by virtue of the DN structure, we have come to an idea that DN gels of complicated shape can be easily prepared from small PAMPS gel particles by forming the second network in the presence of a suitable amount of the cross-linker to unite the particles. To verify this idea, first, we grinded PAMPS bulk gels into particles with several tens micrometres in diameter and dried them (Fig. 5a). The obtained dry PAMPS particles were then soaked in AAm solution to form paste-like solution (Fig. 5b). By polymerizing the AAm of the paste-like solution, DN gels from the PAMPS particles, named ‘P-DN gels’, were formed.
 | ||
| Fig. 5 Free shape formation of robust double network hydrogels starting from PAMPS particles (P-DN gel). (a) PAMPS gel particles (powders) in dried state; (b) swollen PAMPS particles in AAm aqueous solution before gelation; (c) a pair of rabbit meniscus; and (d) artificial meniscus made from P-DN gels. | ||
As presented in Fig. 5c and d, by using this PAMPS particle precursor method, we can synthesize P-DN gels with the same complicated shape of the rabbit meniscus by using complex shaped molds. This method also makes it possible to synthesize the P-DN gels containing versatile particles.
The mechanical strength of P-DN gels strongly depends on the cross-linker concentration of the PAAm network. Fig. 6 presents the fracture energies of P-DN gels and normal DN-gels, as functions of the cross-linker concentration. It was found that the fracture energy of P-DN gels rapidly increases with the increase in the amount of cross-linker used for the polymerization of the PAAm network, and reaches a maximum at 0.01 mol% of the cross-linker, at which concentration the peeling energy of the bonding DN gel sheets also reached their maximum value, as shown in Fig. 2. It is interesting that the P-DN gels become even stronger than the normal DN gels at around 0.01 mol% of the second cross-linker concentration. As the error range of the P-DN gels around 0.01 mol% of the second cross-linker concentration was found to be within several %, so the results are meaningful.
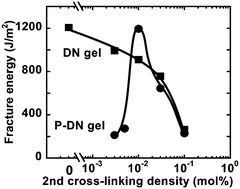 | ||
| Fig. 6 Fracture energy of free-shape P-DN gels and normal DN gels, as functions of the cross-linking density of the second network PAAm. | ||
In order to understand the above phenomenon, let us discuss the results obtained in our previous study on the ‘true-DN gels’.16 Recently, we have developed a method to synthesize the DN gels with no covalent bonding between the two networks, where the residual double bonds of the first PAMPS gels were completely removed by chemical reaction.16 Thus, in this type of the DN gels, named ‘true-DN gels’, the first and the second networks are not inter-cross-linked, i.e., truly independent of each other. It is noted that the fracture energy of the true-DN becomes the largest value, 2200 J m−2 at around 0.01 mol% of the second cross-linker concentration, which corresponds to the concentration for the P-DN gels. It implies that the interface structure among the PAMPS particles in the P-DN gels is similar to the true DN-structure in the true-DN gels. The residual double bonds in the PAMPS particles are not effective for the bridging the particles by inter-cross-linking between the PAMPS particle and the PAAm second network. The added cross-linker for the second network is effective to bridge the particles. Further, the bonding between the gel sheets also becomes the strongest at around 0.01 mol% of the second cross-linker concentration. It implies that the interface structure between the gel sheets is also similar to the true DN-structure.
On the other hand, the effective concentration of the residual double bonds as the cross-linker in normal DN gels was determined as 0.06 mol%.16 This value is quite higher than the optimal value for the true-DN and P-DN gels. Thus, for the normal DN gels, the effective cross-linker concentration becomes 0.06 mol% + 0.01 mol% = 0.07 mol% at the 0.01 mol% of the second cross-linker concentration. It makes the normal DN gels weaker than the P-DN gels even at 0.01 mol%. Additionally, if the P-DN gels have the similar structure to the true-DN gels, the P-DN gels could be improved much stronger than the normal DN gels. The toughening of the P-DN gels is possibly related to the size of the PAMPS particles. The optimal size of the particle could increase the fracture energy of the P-DN gels as high as the true-DN gels. We will continue this work to clarify the effect of the PAMPS particle size on the strength of the P-DN gels.
Here, we like to note that the P-DN gels have the advantages of easy and quick preparation and free-shape forming. While it is difficult to synthesize a DN gel with a desirable complex shape due to the brittleness and size change under swelling of the PAMPS gels, it is easy to handle the paste-like PAMPS particle solutions to molds of various shapes. Also, soaking of AAm monomer in PAMPS gel is a diffusion limited process, which means that the equilibrium soaking time t correlates to the sample size L and the cooperative diffusion constant D (typically, D is ∼10−6 cm2 s−1 for hydrogels32). It implies that 3 mm thick PAMPS gels take about one day to reach the equilibrium state in AAm monomer solution; by scaling down the PAMPS gel to micrometre particles, this process is theoretically shortened by 106 times. In reality, it takes only several minutes to reach swell equilibrium in AAm aqueous solution. Thus, the preparation time can be shortened a lot from a couple of days in total for a normal DN gel to a few hours for P-DN gels. These advantages are extremely important for the large-scale preparation in industry. Also, the P-DN gels will extend the fields of applications. Previously, many studies have been reported about the preparation of functional gels from particles, like iridescent gels, form colloid crystal33 and nanogels.34 Thus, the technology of the P-DN gels will be immediately applied for these fields to prepare high-strength and free-shape functional gels.
Conclusions
In summary, we found that a pair of the first network PAMPS gel sheets is strongly united to each other by introducing the second network PAAm into the system. The strength of peeling energy of the united DN gels became 1200 J m−2, which is comparable to the strength of fracture energy of normal DN gels. The strong bonding due to the DN structure could be achieved if (1) the cross-linker concentration of the second network gels was more than a certain density (0.01 mol%) or (2) the compressive stress on the interface between the first network PAMPS sheets was kept relatively high during the second PAAm polymerization. We also found that this DN-structure technique could be applied to the simple and quick preparation of free-shape particle-DN gels from the small gel particle precursors of the first network PAMPS gels. These findings of both the strong gel–gel bonding and the quick preparation of the high-strength hydrogels, by virtue of the DN-structure technique, no doubt will substantially extend the practical applications of the robust DN hydrogels.Acknowledgements
This research was financially supported by a Grant-in-Aid for the Specially Promoted Research (no. 18002002) from the Ministry of Education, Science, Sports and Culture of Japan. Porous polyethylene (SunfineTM AQ) was provided by Asahi Kasei Chemicals Corporation, Japan. The authors thank Dr W. Wang for her experimental help.References
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- H. Haraguchi and T. Takeshita, Adv. Mater., 2002, 14, 1120–1123 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. Mason, J. Hedrick, Q. Liao, C. Frank, K. Kingsbury and C. Hawker, Chem. Commun., 2006, 2774 RSC.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U.-I. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS.
- M. Guvendiren and K. R. Shull, Soft Matter, 2007, 3, 619–626 RSC.
- Y.-H. Na, T. Kurokawa, Y. Katsuyama, H. Tsukeshiba, J. P. Gong, Y. Osada, S. Okabe, T. Karino and M. Shibayama, Macromolecules, 2004, 37, 5370–5374 CrossRef CAS.
- Y. Tanaka, R. Kuwabara, Y.-H. Na, T. Kurokawa, J. P. Gong and Y. Osada, J. Phys. Chem. B, 2005, 109, 11559–11562 CrossRef CAS.
- Y.-H. Na, Y. Tanaka, Y. Kawauchi, H. Furukawa, T. Sumiyoshi, J. P. Gong and Y. Osada, Macromolecules, 2006, 39, 4641–4645 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- K. Yasuda, N. Kitamura, J. P. Gong, K. Arakaki, H. J. Kwon, S. Onodera, Y. M. Chen, T. Kurokawa, F. Kanaya, Y. Ohmiya and Y. Osada, Macromol. Biosci., 2008, 9, 307–316.
- K. Ito, Curr. Opin. Solid State Mater. Sci., 2010, 14, 28–34 CrossRef CAS.
- K. Haraguchi, M. Ebato and T. Takehisa, Biomacromolecules, 2006, 7, 3267–3275 CrossRef CAS.
- T. Murosaki, T. Noguchi, K. Hashimoto, A. Kakugo, K. Kurokawa, J. Saito, Y. M. Chen, H. Furukawa and J. P. Gong, Biofouling, 2009, 25, 657–666 CrossRef CAS.
- T. Nakajima, H. Furukawa, Y. Tanaka, T. Kurokawa, Y. Osada and J. P. Gong, Macromolecules, 2009, 42, 2184–2189 CrossRef CAS.
- R. Webber, C. Creton, H. R. Brown and J. P. Gong, Macromolecules, 2007, 40, 2919–2927 CrossRef CAS.
- H. R. Brown, Macromolecules, 2007, 40, 3815–3818 CrossRef CAS.
- Y. Tanaka, Europhys. Lett., 2007, 78, 56005 CrossRef , 5 pages.
- T. Tominaga, V. R. Tirumala, E. K. Lin, J. P. Gong and W. L. Wu, J. Phys. Chem. B, 2008, 112, 3903–3909 CrossRef CAS.
- Y. Tanaka, Y. Kawauchi, T. Kurokawa, H. Furukawa, T. Okajima and J. P. Gong, Macromol. Rapid Commun., 2008, 29, 1514–1520 CrossRef CAS.
- H. Furukawa, R. Kuwabara, Y. Tanaka, T. Kurokawa, Y.-H. Na, Y. Osada and J. P. Gong, Macromolecules, 2008, 48, 7173–7178 CrossRef.
- Q. M. Yu, Y. Tanaka, H. Furukawa, T. Kurokawa and J. P. Gong, Macromolecules, 2009, 42, 3852–3855 CrossRef CAS.
- A. G. Peressadko, N. Hosoda and B. N. J. Persson, Phys. Rev. Lett., 2005, 95, 124301 CrossRef CAS , 4 pages.
- B. N. J. Persson, O. Albohr, C. Creton and V. Peveri, J. Chem. Phys., 2004, 120, 8779–8793 CrossRef CAS.
- H. Tsukeshiba, M. Huang, Y.-H. Na, T. Kurokawa, R. Kuwabara, Y. Tanaka, H. Furukawa, Y. Osada and J. P. Gong, J. Phys. Chem. B, 2005, 109, 16304–16309 CrossRef CAS.
- M. Huang, H. Furukawa, Y. Tanaka, T. Nakajima, Y. Osada and J. P. Gong, Macromolecules, 2007, 40, 6658–6664 CrossRef CAS.
- J. P. Gong, G. Kagata and Y. Osada, J. Phys. Chem. B, 1999, 103, 6007–6014 CrossRef CAS.
- C. Tanford, Physical Chemistry of Macromolecules, John Wiley & Sons, Inc., New York, 1961 Search PubMed.
- J. J. Sahlin and N. Peppas, J. Biomater. Sci., Polym. Ed., 1997, 8, 421–436 CrossRef CAS.
- T. Kurokawa, H. Furukawa, W. Wang, Y. Tanaka and J. P. Gong, Acta Biomater., 2010, 6, 1353–1359 CrossRef CAS.
- H. Furukawa, K. Horie, R. Nozaki and M. Okada, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 68, 031406 CrossRef , 14 pages.
- M. Kumada, M. Watanabe and Y. Takeoka, Langmuir, 2006, 22, 4403–4407 CrossRef CAS.
- T. Nishikawa, K. Akiyoshi and J. Sunamoto, J. Am. Chem. Soc., 1996, 118, 6110–6115 CrossRef CAS.
Footnote |
| † Note: it should be noted that in our previous papers,8,9,25,26G was calculated by using a different expression, which is not proper according to the definition. |
| This journal is © The Royal Society of Chemistry 2011 |