DOI:
10.1039/C0PY00139B
(Paper)
Polym. Chem., 2011,
2, 589-594
Metalloenzymatic radical polymerization using alkyl halides as initiators†
Received
3rd May 2010
, Accepted 5th September 2010
First published on 25th September 2010
Introduction
More than a century ago, Buchner discovered that enzymes could catalyze chemical reactions out of the environment of a living cell.1 Since then, in vitro enzymatic synthesis has become a very active field of research both in academia and industry.2 Although the use of enzymes as catalysts for the production of synthetic polymers goes back only two decades,3enzymatic polymerization has become a powerful tool for the mild, selective and sustainable synthesis of materials.4–7 Given the enormous industrial importance of both polyolefins and free radical polymerization processes as well as increasing appreciation of ‘green’ processes, the ability to carry out enzyme mediated radical polymerization of vinyl monomers is particularly appealing.6,8 Horseradish peroxidases (HRP) and laccases have received particular attention in this regard. HRP have been reported to induce conventional free radical polymerization of methacrylic monomers at room temperature in the presence of H2O2 as an oxidant, with9–14 or without15 β-diketones as initiators. Styryl monomers could also be polymerized under free radical conditions with the system HRP/H2O2/β-diketone.14,16 With laccases, polymerization of vinyl monomers such as acrylamide, methyl methacrylate and styrene at 50–80 °C in the absence of any additive, as well as at room temperature in the presence of 2,4-pentanedione has been reported.17–19
In the present work, we show that metalloenzymes such as laccase from the fungus Trametes versicolor (LTV) can induce the radical polymerization of methacrylic monomers in the presence of ascorbic acid and certain alkyl halide (AH) initiators commonly employed in atom transfer radical polymerization20–23 (ATRP). This enabled, for the first time, the introduction of elements of chain-end functionality and architectural control typical of living radical polymerization processes into an enzymatic polymerization.
Experimental section
Materials
Laccase from T. versicolor (LTV, 21.8 units mg−1, Sigma), vitamin C (L-(+)-ascorbic acid, 99+%, Alfa Aesar), L-cysteine (97+%, SAFCTM), ethyl α-bromoisobutyrate (EBiB, assay 98%, Aldrich), 2-bromopropionitrile (2BPN, 97%, Aldrich), methyl chloropropionate (MCP, 97%, Aldrich), ethyl iodoacetate (EIAc, 98%, Aldrich), sodium dodecyl sulfate (SDS, 99%, Sigma-Aldrich) and potassium persulfate (KPS, 99%, Fluka) were used as received. Dry and deoxygenated tetrahydrofuran was withdrawn from a GlassContour Solvent Dispensing System. Water was dispensed from an AquaMAX™ deionized water purification system. Poly(ethylene glycol)methyl ether methacrylate (PEGMA, Mn = 300 g mol−1, Aldrich), methyl methacrylate (MMA, 99%, Aldrich), 2-hydroxyethyl methacrylate (HEMA, 97%, Aldrich), styrene (St, 99+%, Sigma-Aldrich) and divinylbenzene (DVB, tech 80%, Aldrich) were passed through a short column of alumina to remove the inhibitor before use. Poly(ethylene glycol)monomethyl ether bromoisobutyrate (PEG550-BiB)24 and 2-cyano-2-propyl dithiobenzoate (CPDB)25 were prepared according to literature procedures.
In a typical reaction, 0.5 mL of PEGMA were mixed with 2 mL of deionized water to form a homogeneous solution. The mixture was deoxygenated by bubbling argon for ca. 45 min. Laccase (10 mg, ∼200 units) and vitamin C (50 mg) were placed in a Schlenk flask. The flask was sealed with a rubber septum and three vacuum/argon cycles were done. The PEGMA/water mixture was then transferred into the Schlenk flask using an argon-purged syringe. Subsequently, 5 µL of EBiB were syringed into the flask and the resulting mixture was heated to 40 °C in an oil bath.
In a typical reaction, 0.5 mL of PEGMA were mixed with 2 mL of deionized water to form a homogeneous solution. The mixture was deoxygenated by bubbling argon for ca. 45 min. Laccase (10 mg, ∼200 units), vitamin C (50 mg) and L-cysteine (4.1 mg, [EBiB]/[L-Cys] = 1) were placed in a Schlenk flask. The flask was sealed with a rubber septum and three vacuum/argon cycles were done. The PEGMA/water mixture was then transferred into the Schlenk flask using an argon-purged syringe. Subsequently, 5 µL of EBiB were syringed into the flask and the resulting mixture was heated to 40 °C in an oil bath.
In a typical reaction, 0.5 mL of PEGMA were mixed with 1.9 mg of CPDB to form a homogeneous reddish solution. The mixture was deoxygenated by bubbling argon for ca. 30 min. Laccase (10 mg, ∼200 units) and vitamin C (50 mg) were placed in a Schlenk flask. The flask was sealed with a rubber septum and three vacuum/argon cycles were done. First the deoxygenated water (2 mL) and then the deoxygenated PEGMA/CPDB solution were transferred into the Schlenk flask using an argon-purged syringe. Finally, 2.9 µL of 2BPN ([2BPN]/[CPDB] = 4/1) were syringed into the flask and the resulting mixture heated to 40 °C in an oil bath.
In a typical reaction, MMA and deionized water were deoxygenated by bubbling argon for ca. 1 h. Laccase (20 mg, ∼400 units), vitamin C (50 mg) and sodium dodecyl sulfate (SDS, 50 mg) were placed in a single-necked round bottom flask. The flask was sealed with a rubber septum and three vacuum/argon cycles were done. 10 mL of deoxygenated water were then added to the flask using an argon-purged syringe, and a greenish solution was formed. 0.5 mL MMA were then syringed into the flask and the emulsion formed was stirred for about 15 minutes and heated in an oil bath at 40 °C. Subsequently, 5.1 µL of 2BPN were transferred in the flask via a microsyringe to initiate the polymerization. Monomer conversion was determined gravimetrically.
Emulsion polymerization for the synthesis of crosslinked poly(styrene-2-hydroxyethyl methacrylate-divinylbenzene) particles (PSHMD)
The emulsifier-free emulsion copolymerization of styrene (St), 2-hydroxyethyl methacrylate (HEMA) and divinylbenzene (DVB) was carried out under nitrogen in a 50 mL, one-neck round bottom flask equipped with a magnetic stirrer and sealed with a rubber septum. The mixture of monomers (St 0.5 g, HEMA 63.5 mg, DVB 31.2 mg) was introduced into the flask containing 25 mL of deionized water with a stirring rate of 600 rpm. After bubbling nitrogen for 20 minutes, the mixture was heated at 60 °C in an oil bath for 20 minutes. A water solution of potassium persulfate (74.6 mg KPS in 1 mL water) was injected into the mixture to initiate the polymerization. After 20 h, the emulsion was slowly cooled down to ambient temperature. The emulsion was centrifuged at 3500 rpm for 30 minutes to separate the water. The collected solid was then dispersed in methanol under sonication, followed by centrifugation at 3000 rpm for 20 minutes. The washing cycle was repeated for five times in order to remove the unreacted monomers. Next, the solid was dried under vacuum at 45 °C for two days.
Synthesis of (PSHMD) nanoparticles functionalized with 2-bromo-2-methylpropanoate moieties (PSHMD–BiB)
PSHMD (∼120 mg) was dried at 60 °C in a Schlenk flask under vacuum for 2 h in order to remove moisture. After cooling to room temperature, 5 mL of dry THF were placed into a Schlenk flask. The solid was dispersed in THF under sonication for about 40 minutes. While stirring, the flask was immersed in an ice bath and 0.1 mL of triethylamine were added with a microsyringe. Subsequently, 0.1 mL of 2-bromoisobutyryl bromide were added dropwise to the reaction mixture, which was then stirred for 15 hours. The PSHMD–BiB beads were purified by removing THF upon centrifugation (3000 rpm for 20 minutes) and by washing them thoroughly with ethanol (three times), water (twice) and methanol (once). The collected solid was then dried under vacuum at room temperature for 48 hours.
Surface-initiated radical polymerization of PEGMA in the presence of LTV and PSHMD–BiB
PEGMA and deionized water were deoxygenated by bubbling argon for ca. 45 min before use. Laccase (10 mg, ∼200 units) and vitamin C (50 mg) were placed in a Schlenk flask and 50 mg of PSHMD–BiB in another Schlenk flask. The flasks were sealed with a rubber septum and three vacuum/argon cycles were done. The deoxygenated water (2 mL) was then transferred into the Schlenk flask containing laccase and vitamin C using an argon-purged syringe. On the other hand, 0.5 mL of PEGMA were placed in the flask containing PSHMD–BiB. Subsequently, the water solution with laccase/VitC was transferred via an argon-purged syringe into the flask containing the polymer beads and the resulting mixture was heated to 40 °C in an oil bath for 22 hours. The liquid phase was removed upon centrifugation at 4500 rpm for 30 minutes and the resulting solid was dispersed in water under sonication. The centrifugation/sonication cycle was repeated 5 times in order to remove the water-soluble residues. The solid was then dispersed again in water under sonication, followed by low speed centrifugation at 600 rpm for 10 minutes. The opaque bluish slurry was separated from the white color sediment using a syringe. The slurry was then centrifuged at 4500 rpm for 30 minutes to separate the water. Finally, the PEGMA-coated solid was washed thoroughly with ethanol for 5 times and dried under vacuum at room temperature for 48 hours.
Characterization
Monomer conversions were determined by 1H NMR spectroscopy performed on a Bruker UltraShield spectrometer operated at 400 MHz. The number- and weight-average molecular weights (Mn and Mw, respectively) as well as polydispersities (Mw/Mn, PDI) of polymers were measured by size exclusion chromatography (SEC) system equipped with a Waters 515 HPLC pump, a 717plus autosampler, a 2414 refractive-index detector, and the following Styragel® columns arranged in series: guard, HR5E (×2, 4.6 mm ID × 300 mm), HR1 and HR0.5, using THF as an eluent, operated at 0.3 mL min−1 and 40 °C. The instrument was calibrated with poly(methyl methacrylate) standards. The FT-IR spectra were obtained with a Bio-Rad Excalibur FTS-3000MX FTIR spectrometer. The morphology characterization of the polymer particles was carried out on a JEOL JSM-6700 Field Emission Scanning Electron Microscope (FE-SEM). Thermogravimetric analysis (TGA) for polymer samples was performed on TA Instrument SDT2960 Thermogravimetric Analyzer. All tests were conducted under N2 flow (100 mL min−1), using samples of 3–5 mg over a temperature range of 30–800 °C at a scan rate of 10 °C min−1. The particle size of PMMA emulsion was measured using Malvern MS2000 Particle Size Analyzer equipped with a Hydro µP sample dispersion unit.
Results and discussion
LTV belongs to the family of blue multi-copper oxidases, which are used by Nature to catalyze the four-electron reduction of molecular oxygen to water.26LTV is monomeric and its active site contains four Cu(II) centers that are usually classified according to their electron paramagnetic resonance (EPR) features: one type-1 (T1) or blue copper, one type-2 (T2) or normal copper and two type-3 (T3) coppers that are antiferromagnetically coupled and thus EPR silent.26,27 T2 and T3 coppers form a trinuclear cluster in which the metals are arranged in an almost perfect regular triangle and the T3 coppers are connected, both physically and electronically, to the T1 copper by a His-Cys-His tripeptide (Fig. 1). Moreover, T1 and T2 coppers have trigonal coplanar geometries whereas the two T3 coppers a distorted tetrahedral configuration.
 |
| | Fig. 1 Schematic representation of the active site of laccase from T. versicolor. | |
In a first set of experiments, 10 mg of LTV, 50 mg of ascorbic acid (AA), 2 mL of water, 0.5 mL of poly(ethylene glycol) methyl ether methacrylate (PEGMA) and 5 µL of ethyl 2-bromoisobutyrate (EBiB) were reacted at 40 °C. The resulting mixture became rapidly viscous. After 1 h, the monomer conversion was 20%, whereas the number-average molecular weight (Mn) and polydispersity index (PDI) were 272![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da and 2.43, respectively. After 2 h, the viscosity of the mixture was such that syringe sampling was not possible anymore and the resulting polyPEGMA could not be dissolved in THF or DMF. Control experiments showed that the omission of either LTV or AA from the reaction mixture yielded no polymer after 22 h, whereas ca. 2% monomer conversion was obtained in the absence of EBiB in the same reaction time. This indicates that all three species must be present in order to effect significant polymerization. The use of PEG550-BiB, a water-soluble homologue of EBiB, led to similar results implying that, under the conditions used, the solubility of the initiator does not play a crucial role in the process. When 2-bromopropionitrile (2BPN) was employed as the initiator, the polymerization was significantly faster yielding 28% monomer conversion, Mn = 171000 Da and PDI = 1.94 after 30 minutes. Insoluble polyPEGMA was obtained after 1 h reaction time. On the other hand, 15% monomer conversion, Mn = 432000 Da and PDI = 2.27 were obtained after 22 h with ethyl iodoacetate (EIAc). Raising the temperature to 60 °C resulted in a milky reaction mixture, the precipitation of gel-like granules after 1.25 h and only 8% monomer conversion after 22 h. Last but not least, in the presence of methyl 2-chloropropionate (MCP) and 2-cyano-2-propyl dithiobenzoate (CPDB) as initiators, no polymerization took place under comparable experimental conditions. CPDB has been used by Matyjaszewski et al. as an initiator/chain transfer agent in combination with less efficient ATRP catalysts producing well-controlled polymerization of styrene and methyl methacrylate.28
000 Da and 2.43, respectively. After 2 h, the viscosity of the mixture was such that syringe sampling was not possible anymore and the resulting polyPEGMA could not be dissolved in THF or DMF. Control experiments showed that the omission of either LTV or AA from the reaction mixture yielded no polymer after 22 h, whereas ca. 2% monomer conversion was obtained in the absence of EBiB in the same reaction time. This indicates that all three species must be present in order to effect significant polymerization. The use of PEG550-BiB, a water-soluble homologue of EBiB, led to similar results implying that, under the conditions used, the solubility of the initiator does not play a crucial role in the process. When 2-bromopropionitrile (2BPN) was employed as the initiator, the polymerization was significantly faster yielding 28% monomer conversion, Mn = 171000 Da and PDI = 1.94 after 30 minutes. Insoluble polyPEGMA was obtained after 1 h reaction time. On the other hand, 15% monomer conversion, Mn = 432000 Da and PDI = 2.27 were obtained after 22 h with ethyl iodoacetate (EIAc). Raising the temperature to 60 °C resulted in a milky reaction mixture, the precipitation of gel-like granules after 1.25 h and only 8% monomer conversion after 22 h. Last but not least, in the presence of methyl 2-chloropropionate (MCP) and 2-cyano-2-propyl dithiobenzoate (CPDB) as initiators, no polymerization took place under comparable experimental conditions. CPDB has been used by Matyjaszewski et al. as an initiator/chain transfer agent in combination with less efficient ATRP catalysts producing well-controlled polymerization of styrene and methyl methacrylate.28
Interestingly, LTV could also initiate polymerization from an appropriately functionalized surface. Indeed, when crosslinked poly(styrene-2-hydroxyethyl methacrylate-divinylbenzene) (PSHMD) nanoparticles functionalized with 2-bromo-2-methylpropanoate (BiB) moieties were suspended in water, the system LTV/AA induced the polymerization of PEGMA rapidly (Fig. 2). Thermogravimetric analysis (TGA) indicates that polyPEGMA constitutes ∼40% by weight of the resulting particles (Fig. 3), which were also analyzed by means of infrared spectroscopy (Fig. 4). To the best of our knowledge, this is the first example of a surface-initiated metalloenzymatic polymerization of a vinyl monomer. On the basis of this finding, we surmise that the use of multifunctional (macro)initiators might allow for the synthesis of a variety of complex polymeric structures under environmentally benign conditions. Research in this direction is presently underway. Notably, the system LTV/AA/AH was found to be effective also in the emulsion polymerization of hydrophobic monomers such as methyl methacrylate (MMA). More than 90% monomer conversion was obtained in 2 h when 2BPN was used as the initiator, resulting in poly(MMA) particles of ≤100 nm (Fig. 5).
 |
| | Fig. 2
SEM image of PSHMD functionalized with BiB moieties before (left) and after (right) PEGMA polymerization with LTV at 40 °C. | |
 |
| | Fig. 3 First derivative of the TGA trace for PSHMD (bottom), PSHMD functionalized with BiB moieties (middle), and polyPEGMA-coated PSHMD (top) with respect to temperature. | |
 |
| | Fig. 4
FT-IR of PSHMD (bottom), PSHMD functionalized with BiB moieties (middle), and polyPEGMA-coated PSHMD (top). | |
 |
| | Fig. 5
SEM image of polyMMA obtained from the emulsion polymerization of MMA at 40 °C in the presence of LTV and 2BPN. | |
The comparative analysis of the experimental results above with the structural features of LTV suggests a mechanistic picture in which ascorbic acid (E0 = 66 mV vs. SHE at pH = 7)29 reduces at least one copper center (for example the T1 one, for which E0 = 800 mV vs. SHE)27 to Cu(I), which in turn initiates the polymerization by abstracting the halogen atom from the alkyl halides according to an ATRP-type activation process (Scheme 1).
 |
| | Scheme 1 Possible activation/deactivation mechanism involving laccase from T. versicolor and alkyl halides. | |
The fact that no polymerization took place in the absence of AA and that the relative reactivity of the alkyl halides with LTV is comparable with the one found in ATRP with copper complexes30–32 corroborates this hypothesis. However, the low control achieved in the polymerizations indicates that the resulting halogen-containing copper enzyme has limited capability in deactivating the growing polymer chains. A detailed mechanistic investigation is currently underway to address these issues.
Interestingly, the use of a naturally occurring, water-soluble (L)-cysteine (Cys) enabled the regulation of the molecular weights of polyPEGMA and, consequently, the viscosity of the reaction mixture, which also produced beneficial effects on the monomer conversion (>80% with 2BPN as the initiator). Due to the weakness of the S–H bond and the high reactivity of the thiyl radicals, thiols act as chain transfer agents in free radical polymerization.33 The results are summarized in Table S1 (ESI†) and two examples are given in Fig. 6 and 7.
 |
| | Fig. 6 Kinetic plot for the radical polymerization of PEGMA in the presence of LTV for two different cysteine/EBiB molar ratios at 40 °C. Percent monomer conversions are reported in brackets. | |
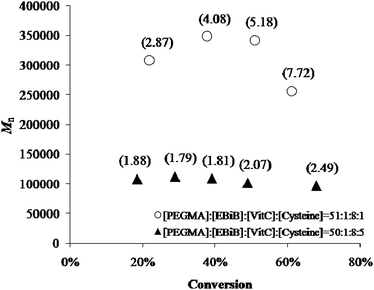 |
| | Fig. 7 Number-average molecular weight of polyPEGMA vs. percent monomer conversion in the presence of LTV for two different cysteine/EBiB molar ratios at 40 °C. Polydispersity indices (Mw/Mn) are reported in brackets. | |
By increasing the number of equivalents of Cys from 1 to 10, the Mn of the corresponding polymers decreased from >300000 to ∼60000 Da and the PDI approached the value of 2, as expected in a polymerization under chain transfer regime. By taking the values of Mn at ∼40% monomer conversion with EBiB as the initiator (Table S2, ESI†), the Mayo-type plot34 in Fig. 8 was obtained, which gives a Ctr = ktr/kp value of 0.023, where ktr is the rate constant for chain transfer and kp the rate constant for monomer propagation.
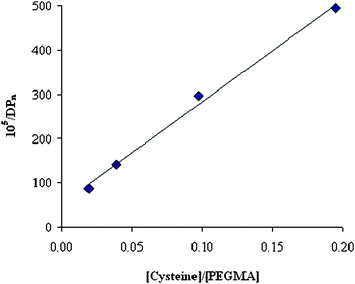 |
| | Fig. 8 Mayo plot for the radical polymerization of PEGMA in the presence of LTV, EBiB and cysteine at 40 °C. | |
On the other hand, when CPDB was used as a chain transfer agent with the system LTV/AA/2BPN/PEGMA in water, a well-controlled, RAFT-type35polymerization took place (Table S3, ESI†). Fig. 9 and 10 report the data at 40 and 50 °C with two different [2BPN]/[CPDB] ratios. In all cases, the semilogarithmic plots of monomer conversion vs. time and the plots of Mnvs. conversion were linear, yet polydispersities as low as 1.35 were obtained. Unsurprisingly, a 10 °C variation in the reaction temperature affected only the ln([M]0/[M]) values and not the molecular weights, whereas Mn turned out to be directly proportional to the ratio [2BPN]/[CPDB].
 |
| | Fig. 9 Kinetic plot for the radical polymerization of PEGMA in the presence of LTV for two different temperatures and CPDB/2BPN molar ratios. | |
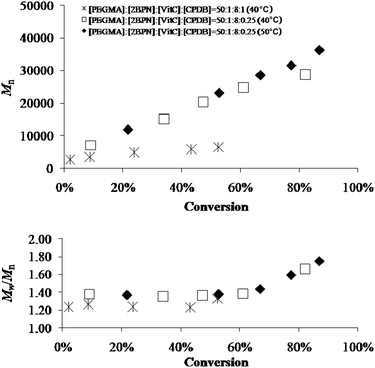 |
| | Fig. 10 Number-average molecular weight of polyPEGMA vs. percent monomer conversion (top) and polydispersity indices (Mw/Mn) of polyPEGMA vs. percent monomer conversion (bottom) in the presence of LTV for two different temperatures and CPDB/2BPN molar ratios. | |
Conclusions
A novel initiation strategy for enzymatic radical polymerization that uses alkyl halides was demonstrated with Laccase from the fungus T. versicolor. The order of reactivity of the alkyl halides resembled that found for copper-catalyzed ATRP, yet the polymerization required the presence of a reducing agent such as ascorbic acid to be effective. Remarkably, the polymerization could also be initiated from a functionalized surface, and it was found to be effective in emulsion as well. The molecular weights of the polymers could be regulated with chain transfer agents such as L-cysteine or 2-cyano-2-propyl dithiobenzoate, the latter resulting in a well-controlled RAFT process. Apart from carrying out fundamental studies on the mechanism with which the polymerization reaction may take place and investigating its application to the sustainable synthesis of new functional materials, we are presently screening a number of enzyme/alkyl (pseudo)halide combinations under various experimental conditions in order to find a feasible catalytic system for the living enzymatic polymerization of vinyl monomers.
The Agency for Science, Technology and Research, Singapore, is gratefully acknowledged for financial support. The authors kindly thank Dr Satyasankar Jana for donation of 2-cyano-2-propyl dithiobenzoate.
References
- E. Buchner, Ber. Dtsch. Chem. Ges., 1897, 30, 117–124 CrossRef CAS.
-
K. Drauz and H. Waldmann, Enzyme Catalysis in Organic Synthesis, Wiley-VCH Verlag GmbH, Weinheim, 2002 Search PubMed.
- J. S. Wallace and C. J. Morrow, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2553–2567 CAS.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS.
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097–2124 CrossRef CAS.
- A. Singh and D. L. Kaplan, Adv. Polym. Sci., 2006, 194, 211–224 CAS.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS.
- R. A. Derango, L.-C. Chiang, R. Dowbenko and J. G. Lasch, Biotechnol. Tech., 1992, 6, 523–526.
- O. Emery, T. Lalot, M. Brigodiot and E. Marechal, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3331–3333 CrossRef CAS.
- D. Teixeira, T. Lalot, M. Brigodiot and E. Marechal, Macromolecules, 1999, 32, 70–72 CrossRef CAS.
- B. Kalra and R. A. Gross, Biomacromolecules, 2000, 1, 501–505 CrossRef CAS.
- A. Durand, T. Lalot, M. Brigodiot and E. Marechal, Polymer, 2001, 42, 5515–5521 CrossRef CAS.
- B. Kalra and R. A. Gross, Green Chem., 2002, 4, 174–178 RSC.
- A. Singh and D. L. Kaplan, J. Polym. Environ., 2002, 10, 85–91 CrossRef CAS.
- H. Uyama, C. Lohavisavapanich, R. Ikeda and S. Kobayashi, Macromolecules, 1998, 31, 554–556 CrossRef CAS.
- A. Singh, D. Ma and D. L. Kaplan, Biomacromolecules, 2000, 1, 592–596 CrossRef CAS.
- R. Ikeda, H. Tanaka, H. Uyama and S. Kobayashi, Macromol. Rapid Commun., 1998, 19, 423–425 CrossRef CAS.
- T. Tsujimoto, H. Uyama and S. Kobayashi, Macromol. Biosci., 2001, 1, 228–232 CrossRef CAS.
- F. Hollmann, Y. Gumulya, C. Tolle, A. Liese and O. Thum, Macromolecules, 2008, 41, 8520–8524 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 Search PubMed.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- N. V. Tsarevsky, T. Sarbu, B. Goebelt and K. Matyjaszewski, Macromolecules, 2002, 35, 6142–6148 CrossRef CAS.
- B. Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2605 CrossRef CAS.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS.
- Y. Kwak, Y. Yamamura and K. Matyjaszewski, Macromol. Chem. Phys., 2010, 211, 493–500 CrossRef CAS.
- H. Borsook and G. Keighley, Proc. Natl. Acad. Sci. U. S. A., 1933, 19, 875–878 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 1858–1863 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS.
- F. di Lena and C. L. L. Chai, Polym. Chem., 2010, 1, 922–930 RSC.
- C. Henrıquez, C. Bueno, E. A. Lissi and M. V. Encinas, Polymer, 2003, 44, 5559–5561 CrossRef CAS.
- F. R. Mayo, J. Am. Chem. Soc., 1943, 65, 2324–2329 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables for the polymerizations of PEGMA with LTV, EBiB or 2BPN and cysteine or CPDB. See DOI: 10.1039/c0py00139b |
| ‡ Present address: Public Research Center “Henri Tudor”, Department of Advanced Materials and Structures, 66 rue de Luxembourg, L-4221 Esch-sur-Alzette, Luxembourg. E-mail: fabio.dilena@tudor.lu |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.