Preparation and rapid degradation of nontoxic biodegradable polyurethanes based on poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) and L-lysine diisocyanate
Zhigao
Wang
,
Lunquan
Yu
,
Mingming
Ding
,
Hong
Tan
*,
Jiehua
Li
and
Qiang
Fu
*
College of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu, China. E-mail: hongtan@scu.edu.cn; qiangfu@scu.edu.cn.; Fax: +86 28 85405402; Tel: +86 28 85460961
First published on 4th November 2010
Abstract
To obtain rapid biodegradable biomaterials, a biodegradable triblock oligomer poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) (PLA-PEG-PLA) was designed and synthesized as a soft segment of polyurethane. Then new nontoxic biodegradable polyurethanes were prepared using the same stoichiometric ratio of PLA-PEG-PLA, L-lysine ethyl ester diisocyanate (LDI), and 1,4-butanediol (BDO). The molecular weights of polyurethanes were controlled by adjusting the polymerization temperature. The resulting polyurethanes were characterized by gel permeation chromatography (GPC), Fourier transform infrared spectroscopy (FTIR), and differential scanning calorimetry (DSC). Furthermore, the biodegradability of the synthesized polyurethanes was evaluated at 37 °C in phosphate buffer solutions (PBS) under different pH values and enzymatic solution at pH 7.4. The results showed that these polyurethanes could be rapidly degraded in PBS and enzymatic solution, as demonstrated by weight loss measurements and scanning electron microscope (SEM) observations. The degradation rates of these polyurethanes were mainly regulated by microphase separation degree, and could be restrained in lower pH value PBS. Moreover, the degradation products did not significantly decrease the pH value of incubation media, which would be useful to improve biocompatibilities of these polyurethanes in vivo. The current work provides a more promising approach to prepare nontoxic biodegradable polyurethanes with rapid degradation rates. These new materials may find potential use for drug delivery systems and magnetic resonance imaging (MRI) contrast agents.
Introduction
Recently, biodegradable synthetic polymers have been widely used in a variety of biomedical applications, including tissue engineering, drug delivery systems, resorbable sutures, and implantable devices.1 Among the families of synthetic polymers, the aliphatic polyesters such as poly(lactic acid) (PLA),2poly(glycolic acid) (PGA),3 poly(lactic-co-glycolide) (PLGA),4 and poly(ortho-esters) (POE),5 have been attractive for these applications due to their degradation by the hydrolysis of ester bonds to form resorbable degradation products.6 However, some biocompatibility concerns of these materials have been raised in recent years. For example, the acidic degradation by-products would cause a decrease of the pH value of the surrounding tissue fluid during in vivo tests.7 Small particles released during degradation might trigger an inflammatory response.8 With development of biodegradable polymer, it is necessary to design and synthesis new nontoxic biodegradable polymers that meet good biocompatibility requirements.Segmented polyurethanes (SPU) have been used as biomaterials due to their excellent physical properties and good biocompatibility.9 The structure of polyurethane can be easily tailored to obtain a broad range of physical, chemical properties, and controllable degradation rates.10,11 However, a major problem is the release of toxic and carcinogenic aromatic diamine from conventional aromatic diisocyanates.
Accordingly, an L-lysine derived diisocyanate (LDI) has been applied to produce nontoxic degradation products.12 Skarja and Woodhouse 13,14 have developed a family of novel biodegradable segmented polyurethanes which are suitable for use in soft tissue applications. The hard segments of these materials were LDI and a phenylalanine diester chain extender, and the soft segments were poly (ε-caprolactone) (PCL) or poly (ethylene oxide) (PEO). Hilborn and co-workers15,16 synthesized highly elastic biodegradable polyurethanes using LDI and biodegradable macrodiols (copolymers of trimethylene carbonate, ε-caprolactone, and D,L-lactic acid) with low degradation rates for long-term tissue engineering scaffolds. Other kinds of biodegradable polyurethanes were also developed with LDI and glycerol for the controlled release of camptothecin by Beckman's group.17,18 However, current studies on these biodegradable polyurethanes are mainly focused on long-term degradation applications, such as tissue engineering and long-term drug delivery. Nevertheless, rapid degradation is a desired property of biodegradable polymer to facilitatte its application in clinical treatment applications, such as rapid drug delivery system19 and magnetic resonance imaging (MRI) contrast agent in biomedical imaging.20 As an amphiphilic block copolymer, polyurethane with hydrophobic and hydrophilic blocks can self-assemble into core-shell structure micelles for drug delivery.21 Amphiphilic polyurethane can also be used as MRI contrast agent because magnetic nanoparticles can be encapsulated inside the hydrophobic core and show good biocompatibility in vivo for its use in MRI.20 They can be easily eliminated from the body due to their rapid degradation.
To prepare new biodegradable polyurethanes for rapid drug delivery system and MRI contrast agent, PEG was introduced to enhance the hydrophilicity and degradability of PLA,22 and natural lysine-derived diisocyanate (LDI) was applied in an attempt to avoid the release of toxic and carcinogenic compounds. A biodegradable triblock oligomer PLA-PEG-PLA was first synthesized. New nontoxic biodegradable polyurethanes with various molecular weights were prepared using PLA-PEG-PLA as soft segment, LDI and BDO as hard segments by adjusting the polymerization temperature. The obtained polyurethanes were fully characterized by GPC, FTIR, and DSC, their in vitrodegradation behavior was also investigated in PBS and enzymatic solution.
Experimental
Materials
Poly(ethylene glycol) (PEG, MW = 1500, obtained from Sinopec Shanghai Gaoqiao Petrochemical Corporation, China) was dehydrated at 100 °C under vacuum for 2 h. L-lactide was purchased from Fushun Tianyuan Biomaterials Co. Ltd (China) and recrystallized three times from ethyl acetate before use. L-lysine ethyl ester diisocyanate (LDI) was synthesized according to reference of Ding et al.,23 and 1,4-butanediol (BDO) (Flaka chemika, Switzerland) was distilled under vacuum. N,N-dimethylacetamide (DMAc) was dried over CaH2 for 2 days at room temperature, distilled under vacuum, and stored in the presence of 4 Å molecular sieves. Lipase from porcine pancreas type II was obtained from Sigma (St. Louis, MO, USA) (lipase activity: 100–400 units/mg protein (using olive oil (30 min incubation)), 30–90 units/mg protein (using triacetin)). The other reagents were used as received.Synthesis of PLA-PEG-PLA
The soft segment PLA-PEG-PLA was synthesized using a ring-opening polymerization,24 as shown in Fig. 1. | ||
| Fig. 1 Synthesis route for PLA-PEG-PLA and polyurethanes. | ||
Briefly, 14.4 g L-lactide and 15.0 g PEG with stannous octoate (0.1 wt %) were added into a round-bottomed flask equipped with a magnetic stirring bar. After degassing at 80 °C for 2 h, the flask was sealed under vacuum and immersed into a thermostatic oil bath at 140 °C for 24 h. The product was dissolved in chloroform, precipitated in anhydrous ethyl ether, and dried under vacuum at 60 °C for 24 h (yield: 80.4%). 1H NMR was employed to characterize the polymer structure, the LA/EG ratio was determined from the integration ratio of resonances due to PEG blocks at 3.64 ppm (–OCH2CH2) and to PLA blocks at 5.20 ppm (–CH), then number-average molecular weight (Mn) was calculated using the following equation:
| DPPEG = (1500 − 18)/44 = 34, DPPLA = DPPEG × (LA/EG)/2, Mn = 1500 + 2 × 72 DPPLA |
Synthesis of polyurethanes based on PLA-PEG-PLA
Polyurethanes were synthesized through a standard two step solution polymerization as shown in Fig. 1. The stoichiometry of the reaction was approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 of hard segment (LDI): soft segment (PLA-PEG-PLA): chain extender (BDO). Firstly, LDI (0.946 g) was added to the stirred DMAc solution of PLA-PEG-PLA (5 g) under a dry nitrogen atmosphere and kept at 75 °C for 2 h. Then BDO (0.179 g) and stannous octoate catalyst (0.1 wt%) were added to the solution and the temperature was raised to 90 °C for 5 h, 100 °C for 1 h. By changing prepolymerization temperature and chain-extension temperature, five PU samples were prepared. Finally, the polymer was precipitated in excess ethyl ether to extract low molecular weight impurities, and dried under vacuum at 60 °C for 24 h to remove residual solvent.
:1:1 of hard segment (LDI): soft segment (PLA-PEG-PLA): chain extender (BDO). Firstly, LDI (0.946 g) was added to the stirred DMAc solution of PLA-PEG-PLA (5 g) under a dry nitrogen atmosphere and kept at 75 °C for 2 h. Then BDO (0.179 g) and stannous octoate catalyst (0.1 wt%) were added to the solution and the temperature was raised to 90 °C for 5 h, 100 °C for 1 h. By changing prepolymerization temperature and chain-extension temperature, five PU samples were prepared. Finally, the polymer was precipitated in excess ethyl ether to extract low molecular weight impurities, and dried under vacuum at 60 °C for 24 h to remove residual solvent.
Characterizations
Nuclear magnetic resonance (1H NMR) spectrum was recorded in CDCl3 with a Varian 400 MHz spectrometer using tetramethylsilane (TMS) as an internal standard. Gel permeation chromatography (GPC) was performed by a PL-GPC 220 (Polymer Laboratory Ltd., England) using N,N-dimethylformamide (DMF)/LiBr as eluent and polymethyl methacrylate (PMMA) as reference. MW of the PMMA standards for GPC system were 2500, 10100, 54500, 93300, 460000, 981000. The sample concentration was 1.000 mg mL−1, and the flow rate was 1.000 mL min−1. Fourier transform infrared (FTIR) spectra were obtained with the Nicolet-560 spectrophotometer between 4000 and 600 cm−1 in the resolution of 4 cm−1. Differential scanning calorimetry (DSC) was carried out using a NETZSCH DSC 204 purged with nitrogen. Data was collected at a heating rate of 10 °C min−1 from −110 °C to 150 °C.Degradation experiments
The PU films (15 mm × 15 mm in size and approximately 0.1 mm thickness) were prepared by solution casting. A 10% (w/v) DMAc solution was cast onto a cover glass, kept in an oven at 50 °C for 24 h, and dried under vacuum at 60 °C for 48 h.In this research, Lipase from porcine pancreas was employed in the enzymatic degradation because its ability to degrade PLA has been documented in previous studies.25 Hydrolytic degradation was carried out in PBS solution (0.1 M PBS with 0.9% NaCl and 0.02% NaN3, pH 7.4, 6 and 5), and enzymatic degradation was carried out in enzymatic solution (0.1 mg mL−1Lipase from porcine pancreas in 0.1 M PBS with 0.9% NaCl and 0.02% NaN3, pH 7.4). Each sample was placed into an individual vial containing 10 mL PBS, and then incubated with shaking at 37 °C to simulate in vivo dynamic tissue environment. The samples were taken out after 6 h, 12 h, 24 h, 48 h and 96 h, rinsed by deionized water, vacuum dried at 60 °C for 24 h, and reweighed to determine weight loss using the following equation:
| Weight loss (%) = (W0 − Wt)/W0 × 100% |
Results and discussion
Synthesis of PLA-PEG-PLA and polyurethanes
Firstly, PLA-PEG-PLA triblock oligomer was synthesized and characterized by 1H NMR. As shown in Fig. 2, the presence of methine (CH) and methyl (CH3) protons in PLA were observed at around 5.20 ppm (c) and 1.58 ppm (a) respectively. The methylene protons in CH2 group of PEG were around 3.64 ppm (b). Based on these assignments, we calculated the LA/EG ratio using the integration ratio of resonances due to PEG blocks at 3.64 ppm (–OCH2CH2) and to PLA blocks at 5.20 ppm (–CH), and then the oligomer Mn was obtained as 2508.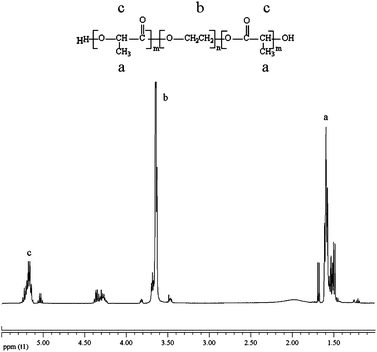 | ||
| Fig. 2 1H NMR spectrum of the PLA-PEG-PLA triblock oligomer. | ||
However, theoretical molecular weight of the oligomer should be 2940 from synthesis prescription, but L-lactide could not be completely polymerized into the oligomer chains due to sublimation of L-lactide at 140 °C, and the molecular weight of polyol obtained was slightly lower than the theoretical value. Herein, it should be noted that a titration method is normally employed to determine the hydroxyl number of polyester or polyether polyol for polyurethane synthesis. However, the hydroxyl number of PLA-PEG-PLA triblock oligomer, which is ester and ether hybrid polyol and contains longer hydrophilic PEG segment, is hardly to be determined using titration because it was facilitately degraded in alkaline solution in our experiment. Comparatively, NMR method is more accurate and has been widely used to determine polyol molecular weight for the synthesis of polyurethane.26,27
Then new nontoxic biodegradable polyurethanes based on LDI, PLA-PEG-PLA, and BDO were prepared. All reagents have been strictly dehydrated in accordance with requirement of general polyurethane preparation before used. By changing the prepolymerization temperature and chain-extension temperature, five PU samples were prepared, which were named as PU1, PU2, PU3, PU4, and PU5 (Table 1).
Polyurethane characterization
The molecular weights of these polyurethanes determined by GPC are listed in Table 1. Their Mn ranged from 3690 to 6240 by varying the reaction temperature. All the samples showed relatively low molecular weights and narrow molecular weight distributions. With the low MWs, these PUs were in an elastic gel state and exhibited poor mechanical properties. However, it is not important for its use in drug delivery and MRI contrast agent. There are two reasons to account for the underestimated molecular weight. One is that the selection of PMMA as reference in GPC measurement is not appropriate for these resulted amphiphilic polyurethanes. Structurally asymmetrical diisocyanate containing primary and secondary isocyanate exhibits different reactivity kinetics is also another possible reason to result in a reduction in molecular weight.22 However, these low molecular weight PUs prepared are useful for the use of drug delivery system and MRI due to their faster degradation. Besides, with various prepolymerization temperatures of 65 °C, 75 °C, 85 °C and a same chain-extension temperature 90 °C, PU2 (75 °C/90 °C) had a higher Mn of 5990 compared to PU1 and PU3. At lower prepolymerization temperature, the polyurethane molecular weight obtained was relatively low probably due to low constant of reaction rate of LDI causing its incomplete reaction at this stage. Similarly, upon increasing the prepolymerization temperature too much, side reactions such as cross-linking of isocyanate also occurred, including the formation of carbamide and allophanate, which resulted in decrease of Mn. Nevertheless, the chain-extension temperature mainly influenced the weight-average molecular weight (Mw) and z-average molecular weight (Mz) (Table 1). These results indicated that the polymerization temperature was one of the key factors affecting the polyurethanes molecular weights.The structures of the polyurethanes were characterized by FTIR, as shown in Fig. 3. FTIR spectra of PU1-PU5 showed characteristic absorption peaks of amino group in urethane at 3340 cm−1 (N–H stretching), 1730 cm−1 peaks were attribute to carbonyl in urethane and ester groups (NHCOO stretching combined with ester COO stretching), and absorption peaks at 1552 cm−1 were ascribed to C–N stretching and N–H out-of-plane bending.28 The absorption peaks of hydrogen bonded carbonyl in urethane groups were located at 1658 cm−1.29 The absorption peaks at 2940 and 2870 cm−1 were corresponded to the symmetric stretching vibration of methyl and methylene.30,31 And 1096, 1134 cm−1 were attributed to ether group (C–O–C) stretching vibration of PEG.30 The NCO peaks in LDI at 2250 cm−1 completely disappeared, clearly implying the successful preparation of PU.
 | ||
| Fig. 3 FTIR spectra of the polyurethanes. | ||
DSC was used to characterize the thermal behavior of the polyurethanes and subsequently to assess the state of microphase separation. Fig. 4 shows the DSC heating curves. It is well known that the thermodynamic incompatibility between hard and soft segments of PU drives a certain microphase separation.9 Because of the low MW, hard and soft segments were not long enough to form highly ordering soft and hard regions, so these PUs showed amorphous state, without any melting endotherm observed in the DSC curves. The glass transition temperatures (Tg) of the soft segments ranged from −33 to −39 °C, which were between the Tgs of PEG (−53 °C) and PLA (60 °C),32 indicating the mixing of hard-soft segment phase in the polyurethane matrix.33 No Tg related to hard segment was observed in DSC curves. This is expected because LDI is structurally asymmetric and also contains methyl ester side chains, which may significantly inhibit chain packing in the hard segment.23 Additionally, it can be observed that Tg of the polyurethanes was closely related to Mn. PU2 and PU5 with relatively high Mn presented high Tg, while PU1, PU3, and PU4 showed low Tg due to polyurethane chains of the lower Mn with high mobility. The decrease of Tg of polyurethanes showed an increased microphase separation between hard and soft segments. Especially, PU4 showed the lowest Tg among these samples, the probable reason is that the hard segment of PU4 with mild cross-linking further enhanced microphase separation of the polyurethane. For PU4, Mz = 20000 was far higher compared to other samples, indicating its viscosity increased,34 which might result from branching or cross-linking.35
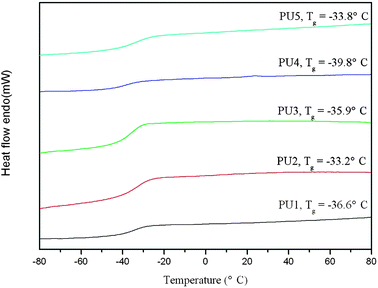 | ||
| Fig. 4 DSC curves of the polyurethanes. | ||
Hydrolytic and enzymatic degradation
Fig. 5 shows the weight loss profiles of the polyurethanes with various molecular weights in enzymatic solution. All five PU samples demonstrated rapid degradation in 96 h, which might be attributed to hydrophilicity of PEG segment, low molecular weight, microphase separation degree, enzyme function and so on. Weight loss initially increased rapidly in first 6 h, and then slowed down, after degradation for 96 h, more than 90% of weight loss was observed for PU1-PU3. This phenomenon is probably attributed to the chains of PUs cleaved into fragments, which were soluble in these aqueous solutions.36 The other reason is that these PUs containg around 49.17% hydrophilic PEG can uptake water, and be easy to swell and dissolve in the aqueous solution.37 Thus, not only did hydrolysis and enzymatic degradation occur, also dissolution of these PUs was considered in mass loss. The dissolvable fragments formed will be helpful to reduce the inflammatory response of vicinity tissues in comparison with insoluble particles. Merely, further work is needed to better understand these degradation products and the dissolvable fragments in the solutions using GPC to measure their molecular weights. As can be seen, weight losses of PU2 and PU3 were increased faster than PU1. Zhang et al. proposed that both lower phase separation degree and Mn led to higher degradation rate of polyurethane.38 This is because the high phase mixing caused more flexibility of polymer chain and weakened the interaction force among the polymer chains, and low initial molecular weight also resulted in the lower polymer chains' interaction, both facilitating water attack, which further increased the rate of hydrolysis.33,38 Additionally, the decreased polymer chains' interaction simultaneously made these hydrophilic polyurethane chains easy to swell and dissolve in water during degradation. PU1 showed lower MW but slower degradation than PU2 and PU3, possibly due to the enhanced interaction force among the polymer chains by the higher degree of microphase separation, which resisted water and enzyme attack. Similar results were also obtained from PU4 and PU5. Here, PU4 and PU5 were degraded more slowly than other samples, and the degradation rate of PU5 exceeded that of PU4 though Mn of PU5 was twice as PU4 (Table1), owing to its lower microphase separation degree. All the results above indicated that their degradation rates might be more affected by microphase separation degree over Mn. | ||
| Fig. 5 Degradation profiles of polyurethanes with various molecular weights in enzymatic degradation. Error bars represent means ± standard deviation for n = 3. | ||
To investigate the effect of enzyme on degradation, comparative test of hydrolytic and enzymatic degradation was carried out. Among the samples, PU2 with relatively high Mn and fast degradation rate was employed in the following degradation experiments. Fig. 6 shows the degradation profiles of PU2 in PBS (pH 7.4) and enzymatic solution, PLA (MW = 5000) is only as a blank for comparison. PLA degradation rate was very slow, even complete degradation needed several weeks to two years.39–41 So in 4 days of degradation, only 13% mass loss was observed for PLA sample. However, PU2 displayed very fast degradation rates (>80%), which were much higher than PLA in both hydrolytic and enzymatic degradation. As can be seen, the enzymatic degradation rate was higher than hydrolytic degradation rate, verifying that Lipase from porcine pancreas can accelerate hydrolysis of these polyurethanes. According to previous studies, the ester linkages in the PLA chain may be expected to be enzymatically hydrolyzable,25 therefore lipase preferably attacked PLA but not the rigid segment.
 | ||
| Fig. 6 Degradation profiles of PU2 and PLA in PBS (pH 7.4) and enzymatic solution. Error bars represent means ± standard deviation for n = 3. | ||
To further investigate surface morphology changes during degradation process, PU films were monitored using SEM. Fig. 7 presents the SEM images of PU2 films during enzymatic degradation (left) and hydrolytic degradation (PBS 7.4) (right). The surface topography of the film before enzymatic degradation was smooth (Fig. 7 (a)). After degradation for 6 h, the surface was eroded markedly, existed holes and cracks (Fig. 7 (b)). The number of cracks and holes became larger as degradation time increased. After 12 h incubation, the polymer film was broken into very small pieces and partially disappeared (Fig. 7 (c)). With degradation time increased to 48 h, film appeared greatly degraded and filamentary structure was observed (Fig. 7 (d)). After 96 h, the film was almost degraded completely, only some small fragments remained (Fig. 7 (e)). Non-enzymatic degradation had a similar phenomenon, but it was slower than enzymatic degradation (Fig. 7 (a′)–(e′)). The SEM results suggested that these polyurethanes could be rapidly degraded, in agreement with weight loss results.
 | ||
| Fig. 7 SEM images of PU2 films during enzymatic degradation at different time (left): (a) original (b) 6 h (c) 12 h (d) 48 h and (e) 96 h, and hydrolytic degradation (PBS 7.4) (right): (a′) original (b′) 6 h (c′) 12 h (d′) 48 h and (e′) 96 h. | ||
The effect of pH value on degradation rate was also evaluated. Fig. 8 shows the degradation profiles of PU2 in PBS at different pH values. The degradation rate was faster in PBS 7.4 solution than that in acid medium (pH 5, 6), which was similar to the observation of Wibullucksanakul et al.42,43 One probable reason is that base promoted the hydrolysis of esters by providing strongly nucleophilic reagent hydroxyl anion (OH−).44,45 The other reason is that degradation fragments contained COOH, which were more soluble in alkaline condition.46
 | ||
| Fig. 8 Degradation profiles of PU2 in PBS at various pH values. Error bars represent means ± standard deviation for n = 3. | ||
In order to further study the effect of degradation products on the pH value, Fig. 9 shows the pH variation of the incubating media during degradation of PU2 in PBS 5, 6, and 7.4, respectively. The pH decreased slightly with time because few acidic degradation products of PLA segment were released, and the alkalinity from amido group of hard segment could possibly neutralize the acidity.36 This result suggested that degradation products from these polyurethanes did not significantly change the pH value of the medium with high weight loss, and then inflammatory responsesin vivo would be reduced.47 Similar results were also observed in the degradation solution of PLA due to its low degradation rate.
 | ||
| Fig. 9 The change in pH values of the incubating media during degradation of PU2 and PLA in PBS 5, 6, and 7.4. Error bars represent means ± standard deviation for n = 3. | ||
Conclusions
In summary, a biodegradable triblock oligomer PLA-PEG-PLA was successfully synthesized as polyurethane soft segment, of which structure and molecular weight were confirmed through 1H NMR. Then new nontoxic biodegradable polyurethanes with various molecular weights have been prepared using the PLA-PEG-PLA, LDI, and BDO by varying polymerization temperature. Their bulk structures were characterized by GPC, FTIR and DSC. These polyurethanes could be rapidly degraded in PBS and enzymatic solution at 37 °C, as verified by weight loss and SEM. Also, their degradation rates were relative to the microphase separation degree, and were probably lowered in acid PBS. Furthermore, the pH value of degradation medium was scarcely changed by degradation products. Thus, these biodegradable polyurethanes will have great potential for biomedical applications such as drug delivery systems48 and MRI contrast agents.49 Further studies are currently being carried out in our group to investigate the micellization behavior and drug delivery characteristics of these polyurethanes.Acknowledgements
Financial supports from National Natural Science Foundation of China (20774061), National 863 project (2008AA03Z304) and New Century Excellent Talents in University (NCET-08-0381) are gratefully acknowledged. We would also like to thank Prof. Qun Gu and Mr. Zongbao Wang at Ningbo Institute of Material Technology & Engineering, Chinese Academy of Sciences for GPC and DSC measurements.Notes and references
- P. Gunatillake, R. Mayadunne and R. Adhikari, Biotechnol. Annu. Rev., 2006, 12, 301 Search PubMed.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569 CrossRef CAS.
- C. P. Barnes, S. A. Sell, E. D. Boland, D. G. Simpson and G. L. Bowlin, Adv. Drug Delivery Rev., 2007, 59, 1413 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS.
- K. P. Andriano, Y. Tabata, Y. Ikada and J. Heller, J. Biomed. Mater. Res., 1999, 48, 602 CrossRef CAS.
- P. Gunatillake and R. Adhikari, Eur. Cell Mater., 2003, 5, 1 CAS.
- C. M. Agrawal and K. A. Athanasiou, J. Biomed. Mater. Res., 1997, 38, 105 CrossRef CAS.
- M. Ara and Y. Imai, Biomaterials, 2002, 23, 2479 CrossRef CAS.
- N. M. K. Lamba, K. A. Woodhouse and S. L. Cooper, Polyurethanes in Biomedical Applications, CRC Press, New York, 1998, pp. 205 Search PubMed.
- X. J. Loh, K. K. Tan, X. Li and J. Li, Biomaterials, 2006, 27, 1841 CrossRef CAS.
- J. J. Guan, K. L. Fujimoto, M. S. Sacks and W. R. Wagner, Biomaterials, 2005, 26, 3961 CrossRef CAS.
- J. Y. Zhang, E. J. Beckman, N. P. Piesco and S. A. Agarwal, Biomaterials, 2000, 21, 1247 CrossRef CAS.
- G. A. Skarja and K. A. Woodhouse, J. Appl. Polym. Sci., 2000, 75, 1522 CrossRef CAS.
- G. A. Skarja and K. A. Woodhouse, J. Biomater. Sci., Polym. Ed., 2001, 12, 851 CrossRef CAS.
- B. Asplund, T. Bowden, T. Mathisen and J. Hilborn, Biomacromolecules, 2007, 8, 905 CrossRef.
- B. Asplund, C. Aulin, T. Bowden, N. Eriksson, T. Mathisen, L. Bjursten and J. Hilborn, J. Biomed. Mater. Res., Part B, 2008, 86, 45 CrossRef.
- W. N. Sivak, I. F. Pollack, S. Petoud, W. C. Zamboni, J. Zhang and E. J. Beckman, Acta Biomater., 2008, 4, 852 CrossRef CAS.
- W. N. Sivak, I. F. Pollack, S. Petoud, W. C. Zamboni, J. Zhang and E. J. Beckman, Acta Biomater., 2008, 4, 1263 CrossRef CAS.
- J. S. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892 CrossRef CAS.
- J. Lu, S. L. Ma, J. Y. Sun, C. C. Xia, C. Liu, Z. Y. Wang, X. N. Zhao, F. B. Gao, Q. Gong, B. Song, X. T. Shuai, H. Ai and Z. W. Gu, Biomaterials, 2009, 30, 2919 CrossRef CAS.
- M. M. Ding, L. J. Zhou, X. T. Fu, H. Tan, J. H. Li and Q. Fu, Soft Matter, 2010, 6, 2087 RSC.
- X. D. Guo, J. P. K. Tan, S. H. Kim, L. J. Zhang, Y. Zhang, J. L. Hedrick, Y. Y. Yang and Y. Qian, Biomaterials, 2009, 30, 6556 CrossRef CAS.
- M. M. Ding, J. H. Li, X. T. Fu, J. Zhou, H. Tan, Q. Gu and Q. Fu, Biomacromolecules, 2009, 10, 2857 CrossRef CAS.
- I. Rashkov, N. Manolova, S. M. Li, J. L. Espartero and M. Vert, Macromolecules, 1996, 29, 50 CrossRef CAS.
- H. Fukuzaki, M. Yoshida, M. Kumakura, T. Mashimo, H. Yuasa, K. Imai and Y. Hidetoshi, Polymer, 1990, 31, 2006 CrossRef CAS.
- W. S. Wang, P. Ping, X. S. Chen and X. B. Jing, Eur. Polym. J., 2006, 42, 1240 CrossRef CAS.
- W. S. Wang, P. Ping, H. J. Yu, X. S. Chen and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5505 CrossRef CAS.
- H. Yeganeh, M. M. Lakouraj and S. Jamshidi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2985 CrossRef CAS.
- X. Jiang, J. H. Li, M. M. Ding, H. Tan, Q. Y. Ling, Y. P. Zhong and Q. Fu, Eur. Polym. J., 2007, 43, 1838 CrossRef CAS.
- H. L. Wang, Y. Zhang, M. Tian, L. F. Zhai, Z. Wei and T. J. Shi, J. Appl. Polym. Sci., 2008, 110, 3985 CrossRef CAS.
- S. A. Guelcher, V. Patel, K. M. Gallagher, S. Connolly, J. E. Didier, J. S. Doctor and J. O. Hollinger, Tissue Eng., 2006, 12, 1247 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley-Interscience, 1998, pp. 227 Search PubMed.
- C. H. Zhang, X. J. Wen, N. R. Vyavahare and T. Boland, Biomaterials, 2008, 29, 3781 CrossRef CAS.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003, pp. 105 Search PubMed.
- S. Subramani, J. Y. Lee, J. H. Kim and I. W. Cheong, Compos. Sci. Technol., 2007, 67, 1561 CrossRef CAS.
- Y. L. Wang, M. N. Huang, Y. F. Luo and Y. G. Li, Polym. Degrad. Stab., 2010, 95, 549 CrossRef CAS.
- R. A. McBath and D. A. Shipp, Polym. Chem., 2010, 1, 860 RSC.
- C. H. Zhang, K. J. Zhao, T. Y. Hu, X. F. Cui, N. Brown and T. Boland, J. Controlled Release, 2008, 131, 128 CrossRef CAS.
- J. E. Bergsma, W. C. Bruijn, F. R. Rozema, R. R. M. Bos and G. Boering, Biomaterials, 1995, 16, 25 CrossRef CAS.
- K. A. Athanasiou, G. G. Niederauer and C. M. Agrawal, Biomaterials, 1996, 17, 93 CrossRef CAS.
- Y. You, S. W. Lee, J. H. Youk, B. Min, S. J. Lee and W. H. Park, Polym. Degrad. Stab., 2005, 90, 441 CrossRef CAS.
- S. Wibullucksanakul, K. Hashimoto and M. Okada, Macromol. Chem. Phys., 1996, 197, 1865 CAS.
- S. Wibullucksanakul, K. Hashimoto and M. Okada, Macromol. Chem. Phys., 1997, 198, 305 CrossRef CAS.
- H. C. Ki and O. O. Park, Polymer, 2001, 42, 1849 CrossRef CAS.
- S. S. Umare and A. S. Chandure, Chem. Eng. J., 2008, 142, 65 CrossRef CAS.
- M. I. Sabir, X. X. Xu and L. Li, J. Mater. Sci., 2009, 44, 5713 CrossRef CAS.
- H. Y. Li and J. Chang, Compos. Sci. Technol., 2005, 65, 2226 CrossRef CAS.
- T. T. Reddy, M. Hadano and A. Takahara, Macromol. Symp., 2006, 242, 41.
- N. Jagielski, S. Sharma, V. Hombach, V. Mailander, V. Rasche and K. Landfester, Macromol. Chem. Phys., 2007, 208, 2229 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |