DOI:
10.1039/C0PY00273A
(Paper)
Polym. Chem., 2011,
2, 432-440
New low-bandgap polymetallaynes of platinum functionalized with a triphenylamine-benzothiadiazole donor–acceptor unit for solar cell applications†‡
Received
27th August 2010
, Accepted 2nd November 2010
First published on 1st December 2010
1. Introduction
The area of sustainable energy is a hot topic in the 21st century. As the most abundant, renewable and clean energy source on the Earth, solar energy is one of the best candidates for solving the worldwide energy crisis.1 Novel materials with lower energy gaps (Eg) need to be developed to improve the coverage of the solar spectrum and consequently improve the efficiency. Organic polymer solar cells (PSCs) offer a promising route for low-cost, environmentally friendly energy generation, provided their efficiencies can be improved to a workable level.2 The bulk heterojunction (BHJ) concept has improved the power conversion efficiency (PCE) of the PSCs significantly by forming a donor–acceptor (D–A) bicontinuous interpenetrated network, which creates large interfacial areas between the polymers and electron acceptors (e.g.fullerene derivatives), thus leading to efficient photoinduced charge separation in a device.3 In this context, low-bandgap conjugated polymers have shown great promise for photoinduced charge generation in BHJ cells to provide solution-processed fabrication for low-cost photovoltaic devices to capture a larger portion of solar energy. A key characteristic of conjugated polymers is the easy modification of their molecular structure and properties by side chain substitution or copolymerization. Different electron donor (D) and electron acceptor (A) moieties with distinct electronic properties have been selected to generate low-bandgap polymers.2 Recently, triphenylamine- and 2,1,3-benzothiadiazole-containing molecules and polymers have attracted much scientific attention because of their intriguing electronic and optical properties and they are widely used in light-emitting diodes (LEDs),4 solar cells5 and two-photon absorption studies.6Aromatic amines such as triphenylamine (TPA) are good hole-transporting chromophores and are commonly used as an electron donor to afford promising photosensitizing materials with improved hole mobility.72,1,3-Benzothiadiazole and its derivatives are known to be strongly fluorescent dyes, which typically exhibit large Stokes shifts.4c, 8 Moreover, an electron-withdrawing 2,1,3-benzothiadiazole unit was used as a useful spacer unit in producing semiconducting organic materials.9
Although recent advances in organic photovoltaics are largely based on conjugated organic polymers, a possible alternative approach for harvesting sunlight to generate electrical power involves putting metals into organic-based polymers.10 Although organometallic donor materials are commonly used in small-molecule solar cells,11 soluble π-conjugated metallopolymers used in high-performance polymer solar cells are still rare to date. Metallopolyynes and their derivatives have recently emerged as one of the most promising photovoltaic polymers due to their high solubility, good thermal stability and diverse structural variability.12 Towards our goal in designing metallopolyyne polymers with low-bandgap π-conjugation for solar cell applications, two new platinum-acetylide polymers consisting of a donor-π-bridge-acceptor-π-bridge-donor (D-π-A-π-D) motif were designed and prepared on the basis of a combination of the 2,1,3-benzothiadiazole unit and two terminal electron-donating TPA groups by π-conjugated spacers to enhance the absorption wavelength and intramolecular charge transfer (ICT) character. BHJ solar cells fabricated from these polymers gave the best power conversion efficiency (PCE) of 1.61% under illumination of an AM 1.5 solar cell simulator. We believe that the present study provides valuable information for the production of new polymer light-harvesting materials for power generation.
2. Experimental
2.1. Materials
Iodine, periodic acid, Pd(OAc)2, CuI, PPh3, K2CO3 and trimethylsilylacetylene were purchased from commercial sources and used as received unless otherwise specified. [6,6]-Phenyl C61-butyric acid methyl ester (PCBM) was purchased from American Dyes. Compounds L1-H,4b, 4cL2-H,4b, 4c, 4dtrans-[Pt(PEt3)2PhCl]13a and trans-[Pt(PBu3)2Cl2]13b were prepared using the methods reported in the literature. Analytical grade solvents were purified by distillation over appropriate drying agents under an inert nitrogen atmosphere prior to use.
2.2. Characterization
The positive-ion fast atom bombardment (FAB) mass spectra were recorded in m-nitrobenzyl alcohol matrices on a Finngin-MAT SSQ710 mass spectrometer. Infrared spectra were recorded on the Nicolet Magna 550 Series II FTIR spectrometer, using KBr pellets for solid state spectroscopy. NMR spectra were measured in deuterated solvents as the lock and reference on a JEOL JNM-EX270 FT NMR system or a Bruker AV 400 MHz FT-NMR spectrometer, with 1H, and 13C NMR chemical shifts quoted relative to Me4Si standard and 31P chemical shifts relative to an 85% H3PO4 external reference. Electronic absorption spectra were obtained with a Hewlett Packard 8453 spectrometer. Optical bandgaps were calculated from the onset of the absorption band. The CV measurements were carried out at a scan rate of 100 mV s−1 using a eDAQ EA161 potentiostat electrochemical interface equipped with a thin film coated ITO covered glass working electrode, a platinum counter electrode and a Ag/AgCl (in 3 M KCl) reference electrode. The solvent in all measurements was deoxygenated MeCN, and the supporting electrolyte was 0.1 M [nBu4N][PF6]. The oxidation and reduction potentials were used to determine the HOMO and LUMO energy levels using the equations EHOMO = [−(Eox(vs.Ag/AgCl) − E(N.H.E.vs.Ag/AgCl)] − 4.50 eV and ELUMO = [–(Ered(vs.Ag/AgCl) − E(N.H.E.vs.Ag/AgCl)] − 4.50 eV, where the potentials for N.H.E. versus vacuum and N.H.E. versusAg/AgCl are 4.50 and −0.22 V, respectively.14 Thin polymer films were deposited on the working electrode by dip coating in chlorobenzene solution (6 mg cm−3). The molecular weights of the polymers were determined by GPC (HP 1050 series HPLC with visible wavelength and fluorescent detectors) using polystyrene standards and thermal analysis was performed with a Perkin-Elmer TGA6 thermal analyzer.
2.3. Synthesis
2.3.1.
L1-I
.
A mixture of L1-H (312 mg, 0.48 mmol), iodine (106 mg, 0.41 mmol) and periodic acid (39 mg, 0.17 mmol) were dissolved in 95% ethanol (25 mL) and CCl4 (25 mL). The reaction mixture was heated to reflux for 10 h. After cooling to room temperature, the solvent was removed and saturated NaHSO3 was added. The precipitate was collected and washed with cold ethanol and dried to provide crude L1-I (390 mg, 0.43 mmol, 90%) as an orange solid. FAB-MS (m/z): 901.9 [M]+. The crude product was difficult to be purified and was used for the Sonogashira reaction without further purification.
2.3.2.
L2-I
.
The compound was prepared in a similar way as for L1-I except that L2-H was used instead. Purple solid. Eluent: hexane/CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v). Yield: 34%. 1H NMR (400 MHz, CDCl3): δ = 8.11 (d, J = 4.0 Hz, 2H, Ar), 7.86 (s, 2H, Ar), 7.57–7.54 (m, 4H, Ar), 7.53–7.51 (m, 4H, Ar), 7.33 (m, 2H, Ar), 7.13–7.11 (m, 4H, Ar), 7.08–7.02 (m, 8H, Ar), 6.87–6.85 (m, 4H, Ar), 2.34 (s, 6H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 152.59, 147.43, 147.10, 145.32, 144.22, 138.11, 133.94, 130.23, 128.69, 128.38, 126.67, 125.65, 125.46, 125.34, 125.19, 123.51, 123.34, 85.00 (Ar), 20.90 (CH3) ppm. FAB-MS (m/z): 1066.0 [M]+.
:2, v/v). Yield: 34%. 1H NMR (400 MHz, CDCl3): δ = 8.11 (d, J = 4.0 Hz, 2H, Ar), 7.86 (s, 2H, Ar), 7.57–7.54 (m, 4H, Ar), 7.53–7.51 (m, 4H, Ar), 7.33 (m, 2H, Ar), 7.13–7.11 (m, 4H, Ar), 7.08–7.02 (m, 8H, Ar), 6.87–6.85 (m, 4H, Ar), 2.34 (s, 6H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 152.59, 147.43, 147.10, 145.32, 144.22, 138.11, 133.94, 130.23, 128.69, 128.38, 126.67, 125.65, 125.46, 125.34, 125.19, 123.51, 123.34, 85.00 (Ar), 20.90 (CH3) ppm. FAB-MS (m/z): 1066.0 [M]+.
2.3.3.
L1-TMS
.
To an ice-cooled mixture of L1-I (174 mg, 0.19 mmol) in freshly distilled triethylamine (10 mL) and CH2Cl2 (10 mL) solution under nitrogen was added Pd(OAc)2 (10 mg), PPh3 (30 mg) and CuI (10 mg). After the solution was stirred for 30 min, trimethylsilylacetylene (0.16 mL, 1.14 mmol) was then added and the suspension was stirred for another 30 min in the ice bath before being warmed to room temperature. After reacting for 30 min at room temperature, the mixture was refluxed for 4 h. The solvents were removed on a rotary evaporatorin vacuo. The crude product was purified by column chromatography on silica gel eluting with hexane–CH2Cl2 (2:1, v/v) to provide L1-TMS (140 mg, 0.17 mmol, 86%) as an orange solid. 1H NMR (400 MHz, CDCl3): δ = 7.88 (d, J = 8.7 Hz, 4H, Ar), 7.74 (s, 2H, Ar), 7.34 (d, J = 8.7 Hz, 4H, Ar), 7.20 (d, J = 8.7 Hz, 4H, Ar), 7.14 (m, 4H, Ar), 7.10–7.03 (m, 8H, Ar), 2.35 (s, 6H, CH3), 0.24 (s, 18H, Si(CH3)3) ppm. 13C NMR (100 MHz, CDCl3): δ = 154.12, 147.85, 147.42, 144.26, 134.12, 132.99, 132.17, 131.56, 130.24, 129.98, 127.53, 125.98, 123.48, 122.48, 116.22 (Ar), 105.39, 93.20 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 20.93 (CH3), 0.08 (Si(CH3)3) ppm. FAB-MS (m/z): 842.5 [M]+. Anal. Calcd for C54H50N4SSi2: C, 76.92; H, 5.98; N, 6.64. Found: C, 77.08; H, 6.15; N, 6.88.
C), 20.93 (CH3), 0.08 (Si(CH3)3) ppm. FAB-MS (m/z): 842.5 [M]+. Anal. Calcd for C54H50N4SSi2: C, 76.92; H, 5.98; N, 6.64. Found: C, 77.08; H, 6.15; N, 6.88.
2.3.4.
L2-TMS
.
This compound was made using the same approach as for L1-TMS but L2-I was used instead. Purple solid. Eluent: hexane/CH2Cl2 (4:1, v/v). Yield: 77%. 1H NMR (400 MHz, CDCl3): δ = 8.11 (m, 2H, Ar), 7.86 (s, 2H, Ar), 7.57–7.55 (m, 4H, Ar), 7.34–7.32 (m, 6H, Ar), 7.13–7.07 (m, 8H, Ar), 7.04–6.98 (m, 8H, Ar), 2.34 (s, 6H, CH3), 0.24 (s, 18H, Si(CH3)3) ppm. 13C NMR (100 MHz, CDCl3): δ = 152.60, 147.75, 147.00, 145.32, 144.17, 138.00, 134.06, 132.99, 130.22, 128.69, 128.59, 126.66, 125.72, 125.67, 125.20, 123.97, 123.38, 122.31, 116.18 (Ar), 105.35, 93.23 (CC), 20.92 (CH3), 0.08 (Si(CH3)3) ppm. FAB-MS (m/z): 1006.2 [M]+. Anal. Calcd for C62H54N4S3Si2: C, 73.91; H, 5.40; N, 5.56. Found: C, 74.12; H, 5.32; N, 5.68.
2.3.5.
L1
.
A mixture of L1-TMS (94 mg, 0.11 mmol) and K2CO3 (138 mg, 1.00 mmol) in a solution mixture of methanol (6 mL) and CH2Cl2 (30 mL), under a nitrogen atmosphere, was stirred at room temperature overnight. The mixture was added to CH2Cl2 (30 mL), washed with water (20 mL) three times and dried over anhydrous Na2SO4. The solvents were removed on a rotary evaporatorin vacuo. The crude product was purified by column chromatography on silica gel eluting with hexane–CH2Cl2 (2:1, v/v) to provide L1 (57 mg, 0.08 mmol, 72%) as an orange solid. 1H NMR (400 MHz, CDCl3): δ = 7.89 (d, J = 8.7 Hz, 4H, Ar), 7.75 (s, 2H, Ar), 7.37–7.35 (m, 4H, Ar), 7.23–7.21 (m, 4H, Ar), 7.16–7.09 (m, 8H, Ar), 7.07–7.05 (m, 4H, Ar), 3.04 (s, 2H, CCH), 2.36 (s, 6H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 154.10, 148.13, 147.36, 144.19, 134.23, 133.11, 132.16, 131.66, 130.28, 130.01, 127.53, 126.07, 123.57, 122.36, 115.01 (Ar), 83.92, 76.30 (CC), 20.94 (CH3) ppm. FAB-MS (m/z): 698.4 [M]+. IR (KBr) (cm−1): ν(CC–H): 3259; ν(CC): 2103. Anal. Calcd for C48H34N4S: C, 82.49; H, 4.90; N, 8.02. Found: C, 82.60; H, 5.07; N, 7.88.
2.3.6.
L2
.
The same method as for L1 was employed to prepare L2 using L2-TMS as the starting material. Purple solid. Eluent: hexane/CH2Cl2 (1:1, v/v). Yield: 96%. 1H NMR (400 MHz, CDCl3): δ = 8.11 (d, J = 4.0 Hz, 2H, Ar), 7.88 (s, 2H, Ar), 7.59–7.57 (m, 4H, Ar), 7.35 (m, 6H, Ar), 7.12–7.09 (m, 8H, Ar), 7.06–7.00 (m, 8H, Ar), 3.04 (s, 2H, CCH), 2.35 (s, 6H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 152.61, 148.05, 146.95, 145.31, 144.12, 138.05, 134.18, 133.12, 130.27, 128.71, 126.70, 125.82, 125.69, 124.08, 123.43, 122.19, 114.97 (Ar), 83.88, 76.30 (CC), 20.93 (CH3) ppm. FAB-MS (m/z): 861.9 [M]+. IR (KBr) (cm−1): ν(CC–H): 3266; ν(CC): 2098. Anal. Calcd for C56H38N4S3: C, 77.93; H, 4.44; N, 6.49. Found: C, 78.12; H, 4.40; N, 6.69.
2.3.7.
P1
.
To a stirred mixture of L1 (50.1 mg, 0.07 mmol) and trans-[Pt(PBu3)2Cl2] (48.1 mg, 0.07 mmol) in freshly distilled triethylamine (20 mL) and CH2Cl2 (20 mL) solution was added CuI (5 mg). The solution was stirred at room temperature for 24 h under a nitrogen atmosphere. The solvents were removed on a rotary evaporatorin vacuo. The residue was redissolved in CH2Cl2 and filtered through a short aluminium oxide column using the same eluent to remove ionic impurities and catalyst residue. After removal of the solvents, the crude product was washed with hexane three times followed by methanol three times and then repeated precipitation from CH2Cl2–hexane (or CH2Cl2–methanol) and drying in vacuo to afford polymer P1 (72 mg, 79%) as a red solid. 1H NMR (400 MHz, CDCl3): δ = 7.85 (d, J = 8.7 Hz, 4H, Ar), 7.72 (s, 2H, Ar), 7.19–7.16 (m, 8H, Ar), 7.10 (m, 8H, Ar), 7.01 (d, J = 8.4 Hz, 4H, Ar), 2.34 (s, 6H, CH3), 2.17–2.14 (m, 12H, PC4H9), 1.62–1.58 (m, 12H, PC4H9), 1.48–1.43 (m, 12H, PC4H9), 0.95–0.91 (m, 18H, PC4H9) ppm. 31P NMR (161 MHz, CDCl3): δ = 2.78 (1JP–Pt = 2350 Hz) ppm. IR (KBr) (cm−1): ν(CC): 2098. Anal. Calcd for C72H86N4P2SPt: C, 66.70; H, 6.69; N, 4.32. Found: C, 66.98; H, 6.86; N, 4.20. GPC (THF): Mw = 9550, Mn = 6840, PDI = 1.40.
2.3.8.
P2
.
Using the above procedure for P1, P2 can be prepared similarly from L2. Purple solid. Yield: 60%. 1H NMR (400 MHz, CDCl3): δ = 8.11 (d, J = 3.6 Hz, 2H, Ar), 7.86 (s, 2H, Ar), 7.54 (d, J = 8.5 Hz, 4H, Ar), 7.31 (d, J = 3.7 Hz, 2H, Ar), 7.17 (d, J = 8.4 Hz, 4H, Ar), 7.11–7.03 (m, 12H, Ar), 6.96 (d, J = 8.4 Hz, 4H, Ar), 2.33 (s, 6H, CH3), 2.17–2.13 (m, 12H, PC4H9), 1.62–1.59 (m, 12H, PC4H9), 1.48–1.43 (m, 12H, PC4H9), 0.95–0.91 (m, 18H, PC4H9) ppm. 31P NMR (161 MHz, CDCl3): δ = 2.79 (1JP–Pt = 2350 Hz) ppm. IR (KBr) (cm−1): ν(CC): 2096. Anal. Calcd for C80H90N4P2S3Pt: C, 65.78; H, 6.21; N, 3.84. Found: C, 65.99; H, 6.12; N, 4.04. GPC (THF): Mw = 10690, Mn = 6540, PDI = 1.63.
2.3.9.
M1
.
To a stirred mixture of L1 (12.8 mg, 0.018 mmol) and trans-[Pt(PEt3)2PhCl] (23.8 mg, 0.042 mmol) in freshly distilled triethylamine (12 mL) and CH2Cl2 (12 mL) was added CuI (2.0 mg). The solution was stirred at room temperature under nitrogen over a period of 24 h. After removal of the solvent, the crude product was purified by column chromatography on silica gel eluting with hexane–CH2Cl2 (1:1, v/v) to give M1 (18.0 mg, 0.010 mmol, 59%) as a red solid. 1H NMR (400 MHz, CDCl3): δ = 7.84 (d, J = 8.8 Hz, 4H, Ar), 7.72 (s, 2H, Ar), 7.34–7.32 (m, 4H, Ar), 7.22–7.16 (m, 8H, Ar), 7.10 (s, 8H, Ar), 7.03–7.00 (m, 4H, Ar), 6.98–6.94 (m 4H, Ar), 6.82–6.78 (m, 2H, Ar), 2.34 (s, 6H, CH3), 1.80–1.73 (m, 24H, PC2H5), 1.14–1.06 (m, 36H, PC2H5) ppm. 13C NMR (100 MHz, CDCl3): δ = 154.18, 148.10, 144.81, 144.16, 139.21, 135.21, 133.03, 132.09, 131.78, 130.18, 129.94, 129.72, 127.27, 125.25, 125.01, 124.47, 124.34, 121.95, 121.16 (Ar), 112.37, 109.73 (CC), 20.88 (CH3), 15.23, 15.06, 14.89, 8.05 (PC2H5) ppm. 31P NMR (161 MHz, CDCl3): δ = 9.86 (1JPt–P = 2626 Hz) ppm. FAB-MS (m/z): 1713.7 [M]+. IR (KBr) (cm−1): ν(CC): 2094. Anal. Calcd for C84H102N4P4SPt2: C, 58.87; H, 6.00; N, 3.27. Found: C, 59.04; H, 5.78; N, 3.21.
2.3.10.
M2
.
Using the above procedure for M1, M2 can be prepared similarly from M2. Purple solid. Eluent: hexane/CH2Cl2 (1:1, v/v). Yield: 82%. 1H NMR (400 MHz, CDCl3): δ = 8.11 (d, J = 4.0 Hz, 2H, Ar), 7.86 (s, 2H, Ar), 7.54–7.52 (m, 4H, Ar), 7.34–7.30 (m, 6H, Ar), 7.22–7.20 (m, 4H, Ar), 7.08–7.04 (m, 12H, Ar), 6.98–6.94 (m, 8H, Ar), 6.82–6.78 (m, 2H, Ar), 2.33 (s, 6H, CH3), 1.80–1.74 (m, 24H, PC2H5), 1.38–1.06 (m, 36H, PC2H5) ppm. 13C NMR (100 MHz, CDCl3): δ = 156.50, 152.61, 147.80, 145.69, 144.73, 144.07, 139.21, 137.58, 135.21, 133.00, 128.68, 127.26, 127.72, 126.46, 125.62, 125.14, 125.01, 124.29, 122.96, 122.44, 121.17 (Ar), 112.48, 109.69 (CC), 20.87 (CH3), 15.23, 15.06, 14.89, 8.05 (PC2H5) ppm. 31P NMR (161 MHz, CDCl3): δ = 9.87 (1JPt–P = 2626 Hz) ppm. FAB-MS (m/z): 1879.7 [M + 1]+. IR (KBr) (cm−1): ν(CC): 2093. Anal. Calcd for C92H106N4P4S3Pt2: C, 58.84; H, 5.69; N, 2.98. Found: C, 58.98; H, 5.84; N, 3.23.
Crystals of L2-H suitable for X-ray diffraction studies were grown by slow evaporation of its solution in a CH2Cl2–hexane mixture at room temperature. X-Ray diffraction data were collected at 173 K using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) on a Bruker Axs SMART 1000 CCD diffractometer. The collected frames were processed with the software SAINT+15 and an absorption correction (SADABS)16 was applied to the collected reflections. The space group was determined from the systematic absences and Laue symmetry check and confirmed by successful refinement of the structure. The structure was solved by the direct method (SHELXTL)17 in conjunction with standard difference Fourier techniques and subsequently refined by full-matrix least-squares analyses on F2. Hydrogen atoms were generated in their idealized positions and all non-hydrogen atoms were refined anisotropically. Crystal data for L2-H: C52H38N4S3, Mw = 815.04, monoclinic, space groupP21/c, a = 11.2608(7), b = 17.823(1), c = 20.189(1) Å, β = 98.705(1)°, V = 4005.4(4) Å3, Z = 4, ρc = 1.352 Mg m−3, μ(Mo-Kα) = 0.229 mm−1, F(000) = 1704, T = 173 K. 24702 reflections measured, of which 9673 were unique (Rint = 0.0372. Final R1 = 0.0575 and wR2 = 0.1505 for 6788 observed reflections with I > 2σ(I). CCDC number 801106.‡
2.5. Fabrication and characterization of bulk heterojunction solar cells
The device structure was ITO/poly(3,4-ethylene-dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS)/polymer:PCBM blend/Al. Indium tin oxide (ITO) coated glass substrates (10 Ω per square) were cleaned by sonication in toluene, acetone, ethanol, and deionized water, dried in an oven, and then cleaned with UV ozone for 300 s. As-received PEDOT:PSS solution was passed through the 0.45 μm filter and spin-coated on patterned ITO substrates at 5000 rpm for 3 min, followed by baking in N2 at 150 °C for 15 min. P1–P2:PCBM (1:4 by weight) active layer was prepared by spin-coating the toluene solution (4 mg mL−1 of metallopolyyne, 16 mg mL−1 of PCBM) at 1000 rpm for 2 min. The substrates were dried at room temperature under low vacuum (vacuum oven) for 1 h and then stored under high vacuum (10−5 to 10−6 Torr) overnight. An Al electrode (100 nm) was evaporated through a shadow mask to define the active area of the devices (2 mm2 circle). All the fabrication procedures (except drying, PEDOT:PSS annealing, and Al deposition) and cell characterization were performed in air. PCE was determined from J–V curve measurement (using a Keithley 2400 sourcemeter) under white light illumination (at 100 mW cm−1). For white light efficiency measurements, an Oriel 66002 solar light simulator with an AM1.5 filter was used. The light intensity was measured by a Molectron Power Max 500D laser power meter.
3. Results and discussion
3.1. Synthesis and characterization
The synthetic routes of the monomers, Pt model compounds and polymers are outlined in Scheme 1. The compounds L1-H and L2-H were synthesized by the Stille coupling of 4,7-dibromo-2,1,3-benzothiadiazole and the appropriate tributylstannyl derivatives with high synthetic yields. Conversion of L1-I and L2-I to their corresponding diethynyl congeners L1 and L2 can be achieved following the typical organic synthetic routes for alkynylation of aromatic halides.18 The two new metallopolyyne polymers (P1 and P2) and their discrete model compound (M1 and M2) were synthesized via the Sonogashira-type dehydrohalogenation reaction between L1 or L2 and a suitable platinum chloride precursor (Scheme 1).19 The feed mole ratio of the platinum precursors and the diethynyl ligands were 1:1 and 2:1 for the polymer and dimer syntheses, respectively. P1 and P2 are thermally and air stable and soluble in common chlorinated hydrocarbons and toluene. P1 and P2 were purified by alumina column chromatography and repeated precipitation, leading to red to purple powders of the polymers in good yield and high purity.
 |
| | Scheme 1 Synthetic routes to the platinum-containing polymers and dinuclear model compounds. | |
The chemical structures of the Pt complexes and polymers were verified by NMR (1H and 31P) and IR spectroscopy. Fig. 1 shows the 1H and 31P NMR spectra of polymers P1 and P2. The signals at 8.11–6.95 ppm are assigned to the resonances of protons on the aryl rings. The single sharp peak at 2.34 ppm belongs to the protons of CH3 groups and the characteristic peaks at 2.17–0.92 ppm are attributed by the PBu units. The single 31P–{1H} NMR signal flanked by platinum satellites for each of the trans-platinum(II) complexes is consistent with a trans geometry of the Pt(PBu3)2 group in a square-planar geometry. The 1JP–Pt values of 2350 Hz for the PBu3 moieties are typical of those for related trans-PtP2 systems.20 All ligands and Pt compounds were subjected to IR measurements in which vibrational frequencies of the acetylenic functional groups were detected. In the IR spectra of the diethynyl ligands, L1 and L2 display IRν(CC) absorption at around 2100 cm−1. The corresponding terminal acetylenic CC–H stretching vibrations occur at 3259 and 3266 cm−1. The CC stretching frequencies for P1 and P2 are located at 2098 and 2096 cm−1, which are lower than those for the free alkynyl precursors, in line with a higher degree of conjugation in the former.21 The molecular structure of L2-H was also confirmed by X-ray crystallography and Fig. 2 depicts a perspective drawing of the molecule.
 |
| | Fig. 2 Molecular structure of L2-H, with the thermal ellipsoids shown at the 25% probability level. | |
The molecular weights of polymers were determined by gel-permeation chromatography (GPC) (see Experimental). The weight-average molecular weights (Mw) of P1 and P2 calibrated against polystyrene standards are 9550 and 10690, respectively. The relatively small polydispersity index (PDI ≈ 1.40–1.63) in molecular weights is consistent with the proposed linear structure from the condensation polymerization. P1 and P2 can possess up to ∼25–28 heterocyclic rings in total along the polymer chain based on number-average molecular weight (Mn). The thermal properties of the polymers were also examined by thermal gravimetric analysis (TGA) under nitrogen (Table 1). P1 and P2 exhibit good thermal stability with the decomposition onsets at ∼355 and 346 °C, respectively, which are higher than those for trans-[–Pt(PBu3)2CCArCC–]n (Ar = C6H4, 300 °C,19b, 19canthrylene, 315 °C,22 oligothienylene, 278–290 °C,23etc.).
Table 1 Photophysical data of the new platinum compounds in CH2Cl2
| |
Absorption (293 K)a |
Emission (293 K) |
Emission (77 K) |
|
λ
abs/nm |
E
g/eVb |
λ
em/nm |
Φ
c
|
τ/ns |
λ
em/nm |
τ/ns |
|
Molar extinction coefficients εmax (104 dm3 mol−1 cm−1) are shown in parentheses.
Optical bandgaps determined from the onset of absorption in solution phases.
Quantum yields determined in CH2Cl2 solution with a CH3CN solution of [Ru(bipy)3](PF6)2 as a standard (Φ = 0.062, λex = 450 nm).29
Not determined.
|
|
M1
|
348 (9.7), 484 (2.3) |
2.09 |
651 |
d
|
0.69 |
613 |
8.11 |
|
M2
|
342 (7.8), 372 (7.2), 542 (4.2) |
1.86 |
704 |
d
|
0.76 |
677 |
2.60 |
|
P1
|
363, 484 |
2.06 |
665 |
0.012 |
0.66 |
617 |
8.92 |
|
P2
|
380, 539 |
1.85 |
716 |
0.018 |
2.84 |
708 |
5.16 |
3.2. Optical properties
The photophysical properties of all new diethynyl ligands and their metal complexes were investigated by UV/Vis and photoluminescence (PL) spectroscopy in CH2Cl2 solution (Table 1). Fig. 3 shows the absorption and PL spectra of the polymers in CH2Cl2 solution. All of the compounds were characterized by two major bands in the absorption spectra. The first one peaking at around 319 nm for L1 and its metal congeners is due to the absorption feature of conjugated arylamine-benzothiadiazole segment. The first peak of L2 and its metal compounds, in the range of 334–369 nm, is attributed to both the absorption of conjugated aromatic segment and π–π* transition in the TPA-thiophene core.24 The second broad absorption peak beyond 400 nm can be assigned to the π–π* transition in the conjugated organic system. As compared to the free alkynes, there is a slight red shift in the absorption wavelength for their corresponding platinum compounds. To enhance π-conjugation relative to L1, M1 and P1, the derivatives L2, M2 and P2 having a thiophene moiety as an additional spacer between the benzothiadiazole ring and the N,N-disubstituted amino group were prepared. The low-bandgap property is attributed to the electron-accepting nature of the benzothiadiazole unit, which causes significant ICT even in the ground state by the combination with the electron-releasing amino groups connected with or without the π-conjugated thiophene spacer. The absorption band of P2 can cover a wide range of 300–700 nm with a correspondingly lower HOMO–LUMO gap of 1.85 eV (versus 2.06 eV for P1).
 |
| | Fig. 3 Normalized absorption and emission spectra of (a) P1 and (b) P2 in CH2Cl2 at 293 K. | |
All of the platinum complexes show weak red to near-infrared emission bands in CH2Cl2 solution at room temperature by direct one-photon excitation (Table 1 and Fig. 3). In general, red luminescent polymers usually show a low emission efficiency because red chromophores are prone to aggregation in the solid state and are highly susceptible to concentration quenching.25P1 shows an emission peak at 665 nm with the lifetime of 0.66 ns at 293 K in dilute solution. Compared to P1, polymer P2 can show a weak emission peak at 716 nm. Due to their similar emission pattern, we consider that the emission features in the metal-free ligand precursors and the Pt compounds should have the same origin. In both cases, no triplet emission was observed which manifests the fate of energy gap law for low-gap polyplatinynes.20d In addition, as described in the literature,12c the fully extended heteroaryl ring in the ligand chromophore greatly reduces the influence of heavy metal ion in P1 and P2 which is mainly responsible for the intersystem crossing and hence the phosphorescence. Therefore, we consider the ligand-dominating singlet excited state instead of the triplet state to contribute to the photoinduced charge separation in the energy conversion for both polymers. All of the Pt compounds undergo a rigidochromic blue shift upon cooling of their solutions to 77 K. The hypsochromic shift observed at low temperature is mainly caused by the solvent reorganization in a fluid solution at 77 K that can stabilize the charge transfer states prior to emission.26 This process is significantly impeded in a rigid matrix at 77 K, and hence luminescence appears at a higher energy. The absence of vibronic progression in the emission profile (both at 293 and at 77 K) suggests mostly a charge-transfer state here but not the ligand-centered π–π* excited state. The observed emission lifetimes (τ) of our Pt compounds at 77 K are shorter than those recorded at room temperature. In these systems, the τ values are generally determined by nonradiative decay rates which usually decreases with lowering temperature. Thus, a lengthening of τ at low temperature would be anticipated.27
The ICT nature is also discussed on the basis of absorption and fluorescent solvatochromism (Fig. 4). The absorption maxima of P1 and P2 exhibit negligible solvatochromic shifts in solvents of increasing polarity, consistent with very small ground-state dipole moments. Contrary to their absorption process, P1 and P2 exhibit pronounced positive solvatochromism in their fluorescence spectra which is diagnostic of the charge-transfer nature of the excited state (Table 2 and Fig. 5), and the finding can be rationalized by the less polar structure of the ground state compared with that of the excited state.28 As a typical example of this phenomenon, with increasing solvent polarity from CCl4, toluene, THF, CHCl3 to CH2Cl2, the λem values of P1 shift significantly to the longer wavelength region by 71 nm (from 594 nm in non-polar CCl4 to 665 nm in polar CH2Cl2). Such solvent-dependent emission in solution is consistent with our expectation for D–A compounds exhibiting significant ICT interaction.
 |
| | Fig. 4 The structure of polymers P1 and P2, with the arrows indicating the expected direction of electron transfer after excitation. | |
Table 2 Charge-transfer absorption and emission wavelength of P1 and P2 in different solvents
|
Solvent
|
P1
|
P2
|
|
λ
max/nm |
λ
em/nm |
λ
max/nm |
λ
em/nm |
|
CCl4 |
489 |
594 |
547 |
649 |
|
Toluene
|
486 |
608 |
544 |
664 |
|
THF
|
483 |
635 |
544 |
694 |
|
CHCl3 |
487 |
651 |
546 |
702 |
|
CH2Cl2 |
489 |
665 |
542 |
715 |
3.3. Electrochemical properties
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels of P1 and P2 were determined using the redox potentials determined from cyclic voltammetry in deoxygenated MeCN, using 0.1 M [nBu4N]BF4 as the supporting electrolyte. From the values of oxidation potential (Eox) and reduction potential (Ered), the HOMO and LUMO levels of P1 and P2 were calculated according to the following equations EHOMO = −(Eox + 4.72) eV and ELUMO = −(Ered + 4.72) eV (where the unit of potential is V versusAg/AgCl).14 The relevant data are collected in Table 3. Both polymers display one irreversible wave at 1.37 and 1.25 eV for P1 and P2, respectively, which can be attributed to the oxidation of electron-rich triarylamino unit. When the thiophene ring is inserted into the triphenylamine-benzothiadiazole moiety in P2, the polymer shows an additional irreversible thienyl oxidation at 1.06 V, which is absent in P1. The electrooxidation of oligothiophene is often irreversible because the electrogenerated cations readily undergo rapid coupling reactions resulting in higher oligomers or polymers.30 The irreversible cathodic wave is attributed to the reduction of the electron-deficient benzothiadiazole moiety. Polymer P2 shows an elevated HOMO energy level (–5.78 eV) relative to P1 (–6.09 eV), indicating that P2 is more electropositive (or has a lower ionization potential) than P1, and a better hole transport ability in P2 can therefore be expected.
Table 3 Electrochemical data and frontier orbital energy levels for P1 and P2
|
Polymer
|
E
ox
/eVa |
E
HOMO/eVb |
E
red/eVc |
E
LUMO/eVd |
|
E
ox
is the oxidation potential.
E
HOMO = −(Eox + 4.72) eV.
E
red is the reduction potential.
E
LUMO = −(Ered + 4.72) eV.
|
|
P1
|
1.37 |
–6.09 |
–1.41 |
–3.31 |
|
P2
|
1.06, 1.25 |
–5.78 |
–1.26 |
–3.46 |
3.4. Photovoltaic properties
Since light-induced intramolecular electron transfer could easily occur from D to A through the π-bridge which favours the photocurrent generation and photoelectronic energy conversion in photovoltaic devices, PSCs were fabricated by using each of P1 and P2 as an electron donor and PCBM as an electron acceptor (Table 4). The hole collection electrode consisted of indium tin oxide (ITO) with a spin-coated PEDOT:PSS, while Al served as the electron collecting electrode. Fig. 6 shows the J–V curves of solar cells with P1–P2:PCBM (1:4, w/w) active layers under simulated AM1.5 solar irradiation. The open circuit voltage (Voc), short-circuit current density (Jsc) and the electrical fill factor (FF) values of the device with P2 as electron donor are 0.78 V, 4.94 mA cm−2 and 0.42, respectively, leading to a PCE of 1.61%. A marked increase in the Jsc and PCE can be observed in P2 relative to P1 at the same blend ratio. It is clear that enhancing the absorption coefficient of the band by increasing the polymer conjugation chain length with additional thienyl rings is an effective way to improve the cell performance. At the same blend ratio of 1:4, the PCE increases in the order P1 < P2 and the light-harvesting ability of P2 can be increased by ∼1.5 times relative to P1 by adding one thienyl ring on both sides of benzothiadiazole. Generally, the amount of absorbed light depends not only on the cut-off absorption wavelength, but also on how intense the absorption is. Comparing model complexes M1 and M2 with different m value, there is an increase in εmax from M1 to M2 (from 2.3 × 104 to 4.2 × 104 dm3 mol−1 cm−1 for the lowest-energy peak), which correlates well with the higher PCE observed for P2 than P1. Therefore, the design of low-bandgap polymers for photovoltaic applications should consider not only lowering the Eg but also increasing the absorption coefficient of the polymer, as well as optimizing the morphology of the blend films. In all diodes, FFs are not very impressive partly because all processing (except PEDOT:PSS annealing and electrode deposition) and measurements have been done in ambient atmosphere which likely results in the presence of traps. We expect FF to improve for fabrication and characterization to be performed in an inert gas environment. Comprehensive study of charge transport and the influence of traps is necessary to further improve FF and overall device performance. Nevertheless, these devices are still not fully optimized. Further studies are needed to optimize the devices based on these materials and the efficient alignment of the energy levels of this type of materials with different acceptors is expected to be a promising strategy for the future advance of organic solar cells.
Table 4 Solar cell performance of the best devices fabricated with P1 and P2
| PSCs |
V
oc/V |
J
sc/mA cm−2 |
FF
|
PCE (%) |
|
P1:PCBM |
0.80 |
4.00 |
0.34 |
1.09 |
|
P2:PCBM |
0.78 |
4.94 |
0.42 |
1.61 |
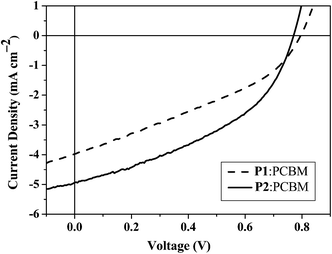 |
| | Fig. 6
J–V curves of solar cells with P1–P2:PCBM (1:4, w/w) active layers under simulated AM1.5 solar irradiation. | |
4. Conclusion
In summary, we have synthesized two stable platinum-acetylide polymers with triphenylamine and 2,1,3-benzothiadiazole as the core components and their photophysical and photovoltaic properties were studied. The solvatochromic effect and low bandgap value are ascribed to ICT through the involvement of strong D–A structural motif in the polymer chain. Adding to this, the extended π-conjugated system including the D and A moieties lowers the excited state energy to let the polymers emit red to near-infrared fluorescence. P2 has a lower Eg than P1 which favours harvesting of more solar photon energy. The expansion of π-electron conjugation is crucial for increasing solar absorption cross-section and the highest PCE of 1.61% was achieved for P2 with a Voc = 0.77 V, Jsc = 4.9 mA cm−2 and FF = 0.39 under illumination of an AM 1.5 solar cell simulator in a 1:4 (P2:PCBM) blend ratio. Our preliminary results indicate the potential solar cell application of metallopolymers P1 and P2. These deeply coloured absorbing polymers are attractive candidates as new functional materials for organometallic photovoltaic technology. However, we need to make more effort to improve the photon-to-electricity conversion efficiency further for practical applications.
Acknowledgements
This work has been supported by the Areas of Excellence Scheme, University Grants Committee (AoE/P-03/08) and a Faculty Research Grant from the Hong Kong Baptist University (FRG2/09-10/II-091). We also thank Drs A. B. Djurišić and Kai-Yin Cheung for their assistance in photovoltaic measurements.
References
- J. A. Turner, Science, 1999, 285, 687–689 CrossRef CAS.
-
(a) Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS;
(b) B. C. Thompson and J. M. J. Frechet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef;
(c) S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef;
(d) K. M. Coakley and M. D. Mcgehee, Chem. Mater., 2004, 16, 4533–4542 CrossRef CAS;
(e) W.-Y. Wong and C.-L. Ho, Acc. Chem. Res., 2010, 43, 1246–1256 CrossRef CAS;
(f) W.-Y. Wong, Macromol. Chem. Phys., 2008, 209, 14–24 CrossRef CAS;
(g) Y. Li and P. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS.
-
(a) G. Yu, J. Gao, J. C. Hummelen, F. A. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS;
(b) N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. A. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS;
(c) J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS;
(d) J. J. M. Halls, K. Pichler, R. H. Friend, S. C. Moratti and A. B. Holmes, Appl. Phys. Lett., 1996, 68, 3120–3122 CrossRef CAS.
-
(a) G. Qian, B. Dai, M. Luo, D. Yu, J. Zhan, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2008, 20, 6208–6216 CrossRef CAS;
(b) J. Liu, L. Chen, S. Shao, Z. Xie, Y. Cheng, Y. Geng, L. Wang, X. Jing and F. Wang, J. Mater. Chem., 2008, 18, 319–327 RSC;
(c) K. R. J. Thomas, J. T. Lin, M. Velusamy, Y.-T. Tao and C.-H. Chuen, Adv. Funct. Mater., 2004, 14, 83–90 CrossRef CAS;
(d) J. Liu, Q. Zhou, Y. Cheng, Y. Geng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Mater., 2005, 17, 2974–2978 CrossRef CAS;
(e) J. Liu, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 101–106 CrossRef.
- W. Li, C. Du, F. Li, Y. Zhou, M. Fahlman, Z. Bo and F. Zhang, Chem. Mater., 2009, 21, 5327–5334 CrossRef CAS.
-
(a) T. Ishi-i, N. Nakamura, T. Mine, S. Imamura, M. Shigeiwa, H. Gorohmaru and S. Maeda, Chem. Lett., 2009, 38, 1042–1043 CrossRef CAS;
(b) S. Kato, T. Matsumoto, M. Shigeiwa, H. Gorohmaru, S. Maeda, T. Ishi-i and S. Mataka, Chem.–Eur. J., 2006, 12, 2303–2317 CrossRef CAS;
(c) S. Kato, T. Matsumoto, T. Ishi-i, T. Thiemann, M. Shigeiwa, H. Gorohmaru, S. Maeda, Y. Yamashitad and S. Mataka, Chem. Commun., 2004, 2342–2343 RSC.
-
(a) W. Zhang, Z. Fang, M. Su, M. Saeys and B. Liu, Macromol. Rapid Commun., 2009, 30, 1533–1537 CrossRef CAS;
(b) Y. Zou, G. Sang, W. Wu, Y. Liu and Y. Li, Synth. Met., 2009, 159, 182–187 CrossRef CAS.
- J. Liu, Q. Zhou, Y. Cheng, Y. Geng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 957–965 CrossRef CAS.
-
(a) C. G. Bangcuyo, U. Evans, M. L. Myrick and U. H. F. Bunz, Macromolecules, 2001, 34, 7592–7594 CrossRef CAS;
(b) Y.-H. Niu, J. Huang and Y. Cao, Adv. Mater., 2003, 15, 807–812 CrossRef CAS.
-
(a) P. D. Harvey and D. Fortin, Coord. Chem. Rev., 1998, 171, 351–354 CrossRef CAS;
(b) W. K. Chan, C. S. Hui, K. Y. K. Man, K. W. Cheng, H. L. Wong, N. Zhu and A. B. Djurišić, Coord. Chem. Rev., 2005, 249, 1351–1359 CrossRef CAS;
(c) W.-Y. Wong, J. Organomet. Chem., 2009, 694, 2644–2647 CrossRef CAS.
-
(a) M. Grätzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef;
(b) M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef CAS;
(c) M. K. Nazeeruddin and M. Gratzel, Struct. Bonding, 2007, 123, 113–175 CAS;
(d) L. M. Goncalves, V. de Zea Bermudez, H. A. Ribeiro and A. M. Mendes, Energy Environ. Sci., 2008, 1, 655–667 RSC.
-
(a) W.-Y. Wong, X. Z. Wang, Z. He, A. B. Djurišić, C. T. Yip, K. Y. Cheung, H. Wang, C. S. K. Mak and W. K. Chan, Nat. Mater., 2007, 6, 521–527 CrossRef CAS;
(b) W.-Y. Wong, X.-Z. Wang, Z. He, K.-K. Chan, A. B. Djurišić, K.-Y. Cheung, C.-T. Yip, A. M.-C. Ng, Y. Y. Xi, C. S. K. Mak and W.-K. Chan, J. Am. Chem. Soc., 2007, 129, 14372–14380 CrossRef CAS;
(c) W.-Y. Wong, W.-C. Chow, K.-Y. Cheung, M.-K. Fung, A. B. Djurišić and W.-K. Chan, J. Organomet. Chem., 2009, 694, 2717–2716 CrossRef CAS;
(d) X.-Z. Wang, W.-Y. Wong, K.-Y. Cheung, M.-K. Fung, A. B. Djurisic and W.-K. Chan, Dalton Trans., 2008, 5484 RSC;
(e) X.-Z. Wang, Q. Wang, L. Yan, W.-Y. Wong, K.-Y. Cheung, A. Ng, A. B. Djurišić and W.-K. Chan, Macromol. Rapid Commun., 2010, 31, 861–867 CrossRef CAS;
(f) F. Guo, Y. G. Kim, J. R. Reynolds and K. S. Schanze, Chem. Commun., 2006, 1887–1889 RSC.
-
(a) G. W. Parshall, Inorg. Synth., 1970, 12, 26–33 CAS;
(b) G. B. Kauffman and L. A. Teterm, Inorg. Synth., 1963, 7, 245–249.
-
(a) J. Hou, Z. Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS;
(b) C. G. Van de Walle and J. Neugebauer, Nature, 2003, 423, 626–628 CrossRef CAS.
-
SAINT, Reference Manual, Siemens Energy and Automation, Madison, WI, 1994–1996 Search PubMed.
-
G. M. Sheldrick, SADABS, Empirical Absorption Correction Program, University of Göttingen, 1997 Search PubMed.
-
G. M. Sheldrick, SHELXTL™, Reference Manual, version 5.1, Siemens, Madison, WI, 1997 Search PubMed.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627–630 CrossRef CAS.
-
(a) W.-Y. Wong, J. Inorg. Organomet. Polym. Mater., 2005, 15, 197–219 CrossRef CAS;
(b) W.-Y. Wong and C.-L. Ho, Coord. Chem. Rev., 2006, 250, 2627–2690 CrossRef CAS;
(c) W.-Y. Wong, Dalton Trans., 2007, 4495–4510 RSC.
-
(a) W.-Y. Wong, G. L. Lu, K. H. Choi and J. X. Shi, Macromolecules, 2002, 35, 3506–3512 CrossRef CAS;
(b) W.-Y. Wong, Coord. Chem. Rev., 2005, 249, 971–997 CrossRef CAS;
(c) W.-Y. Wong, Comments Inorg. Chem., 2005, 26, 39–74 CrossRef CAS;
(d) J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Köhler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412–9417 CrossRef CAS.
-
(a) W.-Y. Wong, C. K. Wong, G. L. Lu, K. W. Cheah, J. X. Shi and Z. Lin, J. Chem. Soc., Dalton Trans., 2002, 4587–4594 RSC;
(b) W.-Y. Wong, C. K. Wong, G. L. Lu, A. W. M. Lee, K. W. Cheah and J. X. Shi, Macromolecules, 2003, 36, 983–990 CrossRef CAS;
(c) J. Lewis, N. J. Long, P. R. Raithby, G. P. Shields, W.-Y. Wong and M. Younus, J. Chem. Soc., Dalton Trans., 1997, 4283–4288 RSC.
- M. S. Khan, M. R. A. Al-Mandhary, M. K. Al-Suti, F. R. Al-Battashi, S. Al-Saadi, B. Ahrens, J. K. Bjernemose, M. F. Mahon, P. R. Raithby, M. Younus, N. Chawdhury, A. Köhler, E. A. Marseglia, E. Tedesco, N. Feeder and S. J. Teat, Dalton Trans., 2004, 2377–2385 RSC.
- N. Chawdhury, A. Köhler, R. H. Friend, W.-Y. Wong, J. Lewis, M. Younus, P. R. Raithby, T. C. Corcoran, M. R. A. Al-Mandhary and M. S. J. Khan, Chem. Phys., 1999, 110, 4963–4970 CAS.
-
(a) G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, D. Ma and Z.-Y. Wang, J. Phys. Chem. C, 2009, 113, 1589–1595 CrossRef CAS;
(b) F. D'Souza, S. Gadde, D.-M. S. Islam, C. A. Wijesinghe, A. L. Schumacher, M. Zandler, Y. Araki and O. Ito, J. Phys. Chem. A, 2007, 111, 8552–8560 CrossRef CAS.
-
(a) C.-T. Chen, Chem. Mater., 2004, 16, 4389–4400 CrossRef CAS;
(b) T. J. Meyer, Pure Appl. Chem., 1986, 58, 1193–1206 CrossRef CAS;
(c) S. D. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949–1960 CrossRef CAS.
- A. B. Tamayo, S. Garon, T. Sajoto, P. I. Djurovich, I. M. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 8723–8732 CrossRef CAS.
- C.-L. Ho, W.-Y. Wong, G.-J. Zhou, B. Yao, Z. Xie and L. Wang, Adv. Funct. Mater., 2007, 17, 2925–2936 CrossRef CAS.
-
(a) Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jové, A. S. Batsanov, J. A. K. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758–2771 CrossRef CAS;
(b) W.-Y. Wong, S.-Y. Poon, M.-F. Lin and W.-K. Wong, Aust. J. Chem., 2007, 60, 915–922 CrossRef CAS;
(c) W.-C. Chow, G.-J. Zhou and W.-Y. Wong, Macromol. Chem. Phys., 2007, 208, 1129–1136 CrossRef CAS.
-
(a) P. Du and R. Eisenberg, Chem. Sci., 2010, 1, 502–506 RSC;
(b) J. M. Calvert, J. V. Caspar, R. A. Binstead, T. D. Westmoreland and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 6620–6627 CrossRef CAS.
-
(a) I. Jestin, P. Frère, N. Mercier, E. Levillain, D. Stievenard and J. Roncali, J. Am. Chem. Soc., 1998, 120, 8150–8158 CrossRef CAS;
(b) A. F. Diaz, J. Crowley, J. Bargon, G. P. Gardini and J. B. Torrance, J. Electroanal. Chem., 1981, 121, 355–361 CAS;
(c) I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281–2305 CrossRef CAS;
(d) R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS;
(e) W.-Y. Wong, G.-L. Lu, K.-F. Ng, K.-H. Choi and Z. Lin, J. Chem. Soc., Dalton Trans., 2001, 3250–3260 RSC.
Footnotes |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| ‡ CCDC reference numbers 801106. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0py00273a |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 
