DOI:
10.1039/C0PY00342E
(Paper)
Polym. Chem., 2011,
2, 441-446
Non-viral siRNA delivery vectors: dendritic molecular transporter and molecular transporter nanovectors for target gene silencing†‡
Received
16th October 2010
, Accepted 31st October 2010
First published on 18th November 2010
Abstract
Viable non-viral delivery vectors have been utilized for the intracellular delivery of siRNA duplexes to result in complete target gene silencing. Biologically active siRNA duplexes were attached to a dendritic molecular transporter (MT) or a molecular transporter-polymeric nanoparticle conjugate (MT-NP) via a thiol exchange reaction utilizing a dithiol pyridyl linker. The MT and the MT-NP delivery systems resulted in significant reduction of the target protein (lamin A/C) investigated for a range of conjugate concentrations. Both non-viral systems have proven effective for the delivery of siRNA into the cytosol of live cells and specifically the development of the MT-NP delivery system provides opportunities for targeted delivery of siRNA into selected cell types.
Introduction
Nucleic acid medicines, such as antisense oligonucelotides (OND) and small interfering RNAs (siRNAs), have tremendous potential as a new class of therapeutic agents. One promising application of nucleic acid (NA) medicines is RNA interference (RNAi), a natural biological process for regulating or silencing gene expression within eukaryotic cells. Discovered by Fire et al. in 1998,1RNAi represents a new approach towards the inhibition of gene expressionin vitro and in vivo and has rapidly emerged as a promising therapeutic target for human diseases, such as cancer,2,3 cardiovascular diseases,4 and other diseases of a genetic etiology.5–7 Current diseases studied in clinical trials for siRNA mediated RNAi include age-related macular degeneration (AMD), diabetic macular edema, hepatitis C, respiratory syncytial virus (RSV) lung infection, pandemic flu, and acute renal failure.8,9 However, a major limitation for in vitro and in vivo therapeutic utilization of siRNAs is the poor cellular uptake and bioavailability of naked siRNA.10–12 Like many nucleic-acid-based therapeutic strategies, the size of a siRNA duplex (∼13 kDa) prevents it from passively diffusing through the cell membrane. Furthermore, the strong anionic charge of the phosphate backbone present in nucleic acids induces electrostatic repulsion from the anionic cell membrane surface thus restricting the therapeutics' entrance into cells.7,13,14 To circumvent these issues research efforts have focused on the development of efficient delivery strategies and has lead to the design of a variety of viral15,16 and non-viral systems for siRNA delivery.10,15,17 While viral systems, or vectors, are the most explored delivery methods for nucleic acids including siRNAs, their clinical application is restricted due to the potential of mutagenicity, oncogensis, host immune responses, as well as the high cost of production.18 Non-viral approaches for siRNA delivery include the use of invasive physical procedures, such as electroporation19 and microinjection,20 and non-invasive systems, like calcium co-precipitation,21 chemical modification of siRNAs,7,22antibodies,23,24 ligand-targeted conjugates,24,25nanoparticles,26,27 complexation with or covalent attachment to cell-penetrating peptides,11,28–31 and complexation with cationic lipids and polymers to form nanoparticles.18,25,32,33 Non-viral vectors are typically preferred over viral vectors due to the ease of their preparation and the low immunogenicity and pathogenicity which allows siRNA therapy with fewer safety concerns.10,34,35 Nevertheless, difficulties still arise in the utilization of some of these non-viral vectors. For example, one difficulty encountered upon direct attachment of negatively charged siRNA with positively charged cell penetrating peptides (CPP) is the neutralization of the positive charges inherent to the CPP, thus preventing the CPP from having interaction with the cell membrane. The neutralization causes the conjugate to loose the ability to bind to the cell surface and become internalized.36,37 Conversely, cationic carriers like lipids and polymers, such as polyethyleneimine (PEI), are based upon charge interactions between positively charged carriers and negatively charged siRNA and often have high toxicity levels due to large amounts of cationic charges that can lead to cell membrane damage and may induce necrosis and apoptosis.5 Additionally, while many non-viral delivery methods avoid the immunogenicity and integration-issues associated with viral techniques, they are not optimized to achieve equal delivery efficiency as that of viral vectors.33,38 Furthermore, the internalization of many of these gene delivery vectors viaendocytosis and delivery to the endosome and lysosome diminishes the efficacy of the delivered siRNA and often requires additional reagents to release the siRNA into the cytosol.26 The need to increase the efficiency of non-viral vectors to make them truly competitive with viral vectors has led to the development and utilization of novel non-viral vectors. Dendritic molecular transporter39,40 and molecular dendritic transporter-nanoparticle vectors41 have recently been developed into versatile intracellular carriers. Previous studies have verified the utilization of the dendritic molecular transporter as a tool for the intracellular delivery of IgG antibodies.40 Additionally, the development and use of a novel nanocarrier that utilizes several hydrolytically stable dendritic molecular transporters in conjunction with a polymeric nanoparticle for the rapid transport of small peptide therapeutics into cells has also been reported.41 Both of these delivery systems employ a pyridinyldithio linker to attach cargo to the vector via an exchange reaction with a sulfhydryl functionality or naturally occurring cysteines on therapeutics to form a disulfide bond. This conjugation approach allows for a facile cleavage of cargo once it has been delivered to the reductive environment of the cytosol in a cell.
In this work, we report the effective delivery of siRNA into human cells utilizing both the dendritic molecular transporter and the molecular dendritic transporter nanoparticle vectors. The efficiency of endogenous lamin A/C knockdown in siRNA-conjugate treated cells is reported and the efficacy of the siRNA-molecular transporter conjugate versus the siRNA-nanoparticle conjugate is compared and analyzed.
Experimental section
Materials
Dendritic molecular transporter40 and molecular transporter-nanoparticle41 vectors were synthesized as previously reported. 5′-thiol modified annealed siRNA, sense (5′-HS-(CH2)6-CUGGACUUCCAGAAGAACA-dTdT-3′) and antisense (5′-UGUUCUUCUGGAAGUCCAG-dTdT-3′), specific for lamin A/C mRNA was purchased from Integrated DNA Technology (IDT) (Coralville, IA). SnakeSkin® Pleated Dialysis Tubing (10,000 MWCO) was obtained from Pierce Biotechnology, Inc. (Rockford, IL). All other reagents were purchased from Sigma-Aldrich, Cellgro, or Invitrogen and used as received. Spectra/Por® Biotech Cellulose Ester (CE) Dialysis Membranes (3,500 MWCO) obtained from Spectrum Laboratories, Inc. (Rancho Dominguez, CA).
siRNA attachment to molecular transporter, 3
A solution of MT, 2, (71.8 μg, 30 nmol) in Dulbecco's phosphate buffered saline solution (DPBS) (pH = 7.4) (6.65 μL) was added to a solution of siRNA, 1, (333.3 μg, 25 nmol) in TE buffer (50 μL) followed by the addition of H2O (200 μL) and TE buffer (200 μL) and the reaction was stirred at room temperature for 5 h and at 4 °C overnight. The reaction was then diluted with H2O and dialyzed against DPBS using SpectraPor® Regenerated Cellulose Ester Dialysis Tubing (3,500 MWCO). Upon completion of dialysis, the aqueous solution was quantitated to give a solution with a siRNA concentration of 10.5 μM (126.2 μg siRNA, 38%).
siRNA attachment to multifunctional nanoparticles, 5
A solution of MFNP, 4, (36.8 μg, 462.9 pmol) in H2O (8.79 μL) was added to a solution of siRNA, 1, (333.3 μg, 25 nmol) in TE buffer (50 μL) followed by the addition of H2O (200 μL) and TE buffer (200 μL) and the reaction was stirred at room temperature for 5 h and at 4 °C overnight. The reaction was then diluted with H2O and dialyzed against DPBS using SnakeSkin® Pleated Dialysis Tubing (10,000 MWCO). Upon completion of dialysis, the aqueous solution was quantitated to give a solution with a siRNA concentration of 9.9 μM (198.2 μg siRNA, 59%).
Lipofectamine-siRNA incubation
Lipofectamine 2000 (Invitrogen) was purchased and used according to the manufacturer's protocol. Briefly, siRNA (100 pmol) was diluted with DMEM (Gibco, 31053) (250 μL). Lipofectamine 2000 (5.0 μL) was diluted with DMEM (Gibco, 31053) (250 μL) and incubated for 5 min at room temperature. The diluted siRNA was then combined with the Lipofectamine 2000 solution and the mixture incubated at room temperature for 20 min. The lipofectamine-siRNA complexes were then added to dishes of HeLa cells under the same conditions as below.
siRNA transfection
The RNAi study was aimed at targeting lamin A/C by the use of double-stranded RNA with sense sequence (5′-HS-(CH2)6-CUGGACUUCCAGAAGAACA-dTdT-3′) and antisense sequence (5′-UGUUCUUCUGGAAGUCCAG-dTdT-3′).
Methods
Cleavage of disulfide on siRNA.
Double stranded (ds)-siRNA (50 nmol) was dissolved in a 10 mM solution of dithiothreitol (DTT) (50 μL) in Tris-EDTA buffer (TE buffer) (Cellgro, Herndon, VA) (pH = 7.8) and was reacted for 30 min. To remove DTT, the ds-siRNA solution was diluted with an additional 50 μL of TE buffer and was then extracted with ethyl acetate (200 μl, 4×) to yield ds-siRNA with a free thiol modification on the 5′ sense strand, 1.
Cell culture and siRNA transfection.
HeLa cells were grown in Dulbecco's modified eagle's medium (DMEM, Sigma-Aldrich, D6046) supplemented with 10% fetal bovine serum (ATCC, 30–2020) and 1% antibiotic-antimycotic (Invitrogen, 15240) at 37 °C with 5% CO2 in a 95% humidity incubator.
Confocal microscopy
.
HeLa cells were plated on uncoated, 14 mm diameter Microwell, No. 1.5 MatTek dishes at a density of 3 × 105cells per well in medium. On the day of experiments, cells were washed three times with DPBS and then incubated with serum and phenol red free, high glucose DMEM (Gibco, 31053), NP, MT, control siRNA (free siRNA), Lipofectamine 2000-siRNA, MT-Lamin A/C siRNA, NP-Lamin A/C siRNA, or NP-Firefly Luciferase siRNA conjugates ([NP] ∼ 4.3 nM or 17.7 nM, [MT] ∼ 250 nM or 1000 nM and [siRNA] ∼ 250–1000 nM) for 6 h at 37 °C in a 5% CO2 environment. After incubation, the complexes were replaced with 2000 μL fresh DMEM with 10% FBS and allowed to incubate for an addition 24 h for RNAi to take effect. For confocal microscopy analysis, cells were fixed on dishes and then incubated with primary antibody to lamin A + C (Abcam, ab8984) (0.01 mg/mL) and then with a fluorescently labeled secondary antibody (Abcam, ab6785) (2 μg/mL). Confocal microscopy was performed using a Zeiss Inverted LSM 510 Meta laser scanning confocal microscope, equipped with an argon laser, a 543 nm HeNe laser, a Plan-Neofluar 40×/1.3 oil lens and a Plan-Apochromat 63×/1.4 oil lens. Samples were analyzed using two channel confocal laser scanning microscopy to obtain a DIC image combined with a FITC fluorescence image.
Flow cytometry
.
HeLa cells were plated onto six-well culture plates at 1 × 106cells/well and allowed to incubate in DMEM containing 10% FBS at 37 °C and 5% CO2 for 24 h. The medium was removed and the cells washed three times with DPBS and subsequently transfected with the appropriate treatment concentrations for 6 h at 37 °C. The treatments were then replaced with cell culture media and the cells incubated for an additional 24 h. After incubation, the cells were washed three times with DPBS, trypsinized, pelleted, washed in DPBS containing 2% BSA, pelleted, and resuspended in methanol for 10 min. The cells were then pelleted, resuspended in a primary antibody solution in 10% BSA in DPBS and incubated at 4 °C overnight. The cells were again pelleted, washed with 2% BSA in DPBS, and resuspended in a secondary antibody solution in 10% BSA in DPBS and incubated at room temperature for 1 h. The cells were pelleted, washed with 2% BSA in DPBS, and resuspended in DPBS for fluorescent activated cell sorting (FACS) analysis. The fluorescence intensity of each cell was detected and acquired using a 5-laser BD (Becton, Dickinson and Company, Franklin Lakes, NJ) LSRII flow cytometer, equipped with a 535 nm laser for the detection of phycoerythring (PE) dye and FACSDiva™ software (BD Biosciences) for data analysis. The positive fluorescence level was established by visual inspection of the histogram of untreated cells such that a small percentage appeared in the positive region. For each sample, fluorescence was acquired on 50,000 cells. The percent silencing reported was derived from the difference between the fluorescence intensities of the untreated control cells and that of the samples that were treated with siRNA conjugates.
Instrumentation.
The oligonucleotide concentration of the siRNA-MT and siRNA-NP conjugates was measured viaUV-Vis using a NanoDrop 1000 Spectrophotometer using the RNA nucleic acid setting and measuring a 2 μL sample. The software converts absorbency to concentration using an extinction coefficient of 40.
Results and discussion
As previously reported, we have designed, synthesized, and incorporated a dendritic molecular transporter (MT)39,40 into a molecular dendritic transporter-nanoparticle vector (MT-NP)41 for the intracellular delivery of therapeutics. Having established in vitro uptake of the molecular transporter, molecular transporter antibody and molecular dendritic transporter-nanoparticle peptide vectors, we extended the utility of these non-viral vectors to transporting and delivering oligonucleotides, such as siRNA. For these vectors, therapeutic cargo must contain a sulfhydryl functionality for the exchange reaction to the MT or MT-NP via a pyridinyldithio linker. In here, the formation of bioconjugates with siRNA and the MT and MT-NP vectors was pursued with 5′-thiol modified siRNA. As a model system, we employed a siRNA known to silence the gene encoding lamin A/C protein present inside the nuclear lamina of cells.42 We began the synthesis of the siRNA conjugates with the revelation of a sulfhydryl functionality on the siRNA by removing the thiol protecting group on the sense strand with dithiothreitol (DTT) in Tris-EDTA buffer (TE). The deprotected siRNA, 2, was purified by extracting the DTT using ethyl acetate followed by directly reacting with the molecular transporter (MT), 3, or the molecular dendritic transporter-nanoparticle (MT-NP), 4, in a DPBS/TE mixture to afford the siRNA bioconjugates 3 and 5, respectively (Schemes 1 and 2, respectively).
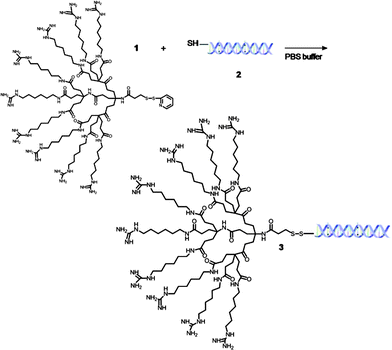 |
| | Scheme 1 Formation of siRNA-MT bioconjugate 3. | |
 |
| | Scheme 2 Formation of siRNA-MT-NP bioconjugate 5. | |
The conjugation reactions were subsequently purified by dialysis against DPBS to remove any unreacted materials. The conjugates were analyzed by UV-Vis, employing the nucleic acid RNA-40 setting on a NanoDrop 1000 Spectrophotometer, to determine the siRNA concentration of each solution. The siRNA concentration of the siRNA-MT conjugate, 3, was found to be 10.5 μM. Also, the siRNA concentration of the siRNA-NP conjugate, 5, was found to be 9.9 μM translating to ∼74% siRNA attachment to the pyridinyldithio linkers on the MT-NP carriers. For in vitro studies, HeLa cells were incubated for 6 h with siRNA-MT or siRNA-MT-NP conjugates using siRNA concentrations of 250–1000 nM and fixed 24 h later. The cells were then stained with a primary mouse monoclonal antibody to lamin A/C and a FITC labeled secondary goat polyclonal antibody to mouse IgG. For comparison, we also employed a commercial transfection agent, Lipofectamine 2000 (LF2000) for siRNA delivery. Confocal analysis of untreated cells exhibited staining of the lamin A/C protein in the nuclear lamina of the imaged cells (Fig. 1, a, see also Supplementary Information). Cells treated with siRNA-LF2000, prepared according to manufacturer's protocol, showed a moderate knockdown of lamin A/C protein as is indicated by the decrease in fluorescent labeling on the nuclear lamina of the cells (Fig. 1, a). However, confocal images revealed more significant reductions in lamin A/C protein expression in cells treated with the non-viral vector conjugates (Fig. 1 and 2) relative to untreated and siRNA-LF2000 treated control cells. Cells treated with siRNA-MT conjugates exhibited a reduction in the amount of lamin A/C protein produced; specifically, it was observed that increasing the conjugate concentration from 250 nM siRNA to 750 nM siRNA resulted in a further discernible reduction of lamin A/C expression (Fig. 1). Confocal imaging of MT treated cells still showed production and labeling of lamin A/C protein, verifying that the MT carrier does not have an effect on the target protein production (Fig. 1, D). Fluorescent activated cell sorting (FACS) analysis of siRNA-MT treated cells supported the linear trend in the potency of RNAi as was seen in the confocal images; with the percentage of silencing increasing with the siRNA-MT concentration and higher concentrations of the conjugate had a greater silencing effect than that of LF2000 (Fig. 3).
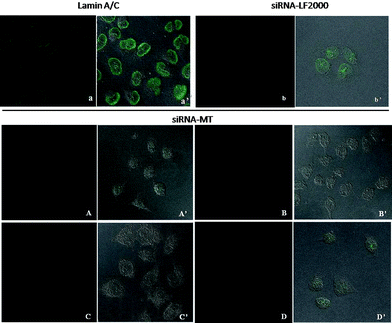 |
| | Fig. 1 Top. Confocal imaging of untreated and siRNA-LF2000 treated HeLa cells: a, fluorescent image of untreated Hela cells imaged for labeled lamin A/C protein; a′, DIC image of HeLa cells of a; b, fluorescent image of siRNA-LF2000 treated HeLa cells; b′, DIC image of HeLa cells of b. Bottom: Confocal imaging of siRNA-MT (3) treated HeLa cells: treated with 250 nM siRNA-MT (A) and DIC image of HeLa cells of A (A′); treated with 500 nM siRNA-MT (B) and DIC image of HeLa of B (B′); treated with 750 nM siRNA-MT (C) and DIC image of C (C′); treated with 1000nM MT (1) control and DIC image of D (D′). | |
 |
| | Fig. 2 Confocal imaging of siRNA-MT-NP (5) treated HeLa cells: fluorescent image of cells treated with 250 nM siRNA-MT-NP (a) and DIC image of a (a′); 500 nM siRNA-MT-NP (b) and DIC image of b, (b′); 750 nM siRNA-MT-NP (c) and DIC image of c (c′); image of control (d) treated with 1000 nM MT-NP (4) control and DIC image of HeLa cells (d′). | |
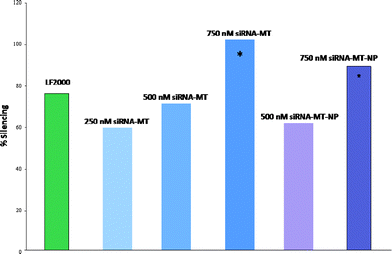 |
| | Fig. 3 Silencing efficacy of LF 2000 (green), 250 nM siRNA-MT (light blue), 500 nM siRNA-MT (Tennessee Titans blue), 750 nM siRNA-MT (dark blue), 500 nM siRNA-MT-NP (violet), 750 nM siRNA-MT-NP (purple). | |
Likewise, confocal imaging of siRNA-MT-NP treated cells also exhibited an increase in target gene knockdown as related to the increase in conjugate concentration (Fig. 2). Again, cells treated with MT-NP showed no change in the production of lamin A/C protein, verifying that the MT-NP carriers also have no apparent effect on the target protein production (Fig. 2, d). Additionally, flow cytometry data from siRNA-MT-NP treated cells report a similar linear trend to that of siRNA-MT treated cells. The data correlates that the percentage of silencing increased with the increase in siRNA concentration and higher conjugate concentrations had greater RNAi potency as compared to LF2000 (Fig. 3). It should be noted that at a siRNA concentration of 750 nM, the siRNA-MT-NP conjugate caused an 89% silencing and the siRNA-MT conjugate had a 100% silencing effect or a complete knockdown of the lamin A/C protein in treated cells (Fig. 3, indicated by *) verifying the efficiency of the novel non-viral vectors. As previously mentioned lamin A/C is inside the nuclear lamina of cells and represents a very small percentage of the cell volume, which can cause complications in certain analytical techniques. For example, confocal microscopy was able to clearly record the fluorescent labeling of the lamin A/C protein; however, flow cytometry analysis of these labeled cells was more complex. FACS exhibited issues with the detection of the labeling within the cells due to the small amount of lamin A/C protein and the resulting small quantity of fluorescent labeling. The small change in signal between cell background and the lamin A/C label lead to a higher percent error for flow cytometry measurements as compared to confocal images. This fact can makes it difficult to discern the silencing effects of the conjugates. Therefore, the FACS data are presented as supporting evidence to further prove the silencing effects that were observed in the confocal images. Both confocal and FACS analysis indicate that siRNA-MT and siRNA-MT-NP conjugates have similar transfection efficacies; however, MT vectors can only have either a therapeutic or a targeting unit attached to each transporter. MT-NPs are more versatile vectors because of their ability to attach targeting units to the nanoparticle backbone in addition to the therapeutic payload. These targeting units can be utilized to create a more selective oligonucleotide delivery tool when a targeted delivery is desired. Moreover, experimentation treating cells with naked siRNA did not exhibit a significant reduction in lamin A/C protein expression which confirms the necessity of a suitable siRNA delivery vector for therapeutic intracellular delivery. Additionally, cells treated with a different siRNA-MT-NP conjugate, where the siRNA is known to silence the gene encoding firefly luciferase protein, did not exhibit a significant reduction in lamin A/C protein, verifying the target gene specificity of the novel delivery system.
Conclusion
In summary, we have prepared siRNA oligonucelotides-molecular transporter MT and a molecular dendritic transporter-nanoparticle vector MT-NP via a cleavable disulfide linkage. The facile attachment of sulfhydryl modified siRNA to the vectors is carried out in aqueous conditions and the MT-NP carrier exhibited ∼74% attachment of siRNA duplexes to available attachment sites on the particle. In vitro studies of these novel bioconjugates have demonstrated highly efficient delivery of siRNAvia MT and MT-NP vectors. Treated cells demonstrated a significant decrease in the fluorescent labeling of the target gene lamin A/C as compared to untreated and LF2000 treated cells. Furthermore, FACS analysis of transfected cells supported the confocal data and reported a linear increase in the silencing efficacy as the concentration of the conjugate increased. A siRNA concentration of 750 nM resulted in 100% (siRNA-MT) or 89% (siRNA-MT-NP) silencing of lamin A/C and these effects present a more potent RNAi functionality than the widely used commercial transfection agent, Lipofectamine 2000. The MT and MT-NP delivery systems provide a viable non-viral alternative for oligonucleotide or small molecule intracellular delivery. Additional targeting units can be incorporated into the MT-NP system to create a highly targeted and efficient siRNA carrier.
Acknowledgements
S.K.H. and E.H. gratefully acknowledge financial support from the CAREER program of the National Science Foundation under Award CH-0645737. Confocal imaging was performed through the use of the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). Flow cytometry experiments were performed in the VMC Flow Cytometry Shared Resource, supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). The authors also thank Vanderbilt University (VINSE, Vanderbilt Institute of Nanoscale Science and Engineering) for supporting this research.
Notes and references
- A. Fire, S. Q. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS.
- G. R. Devi, Cancer Gene Therapy, 2006, 13, 819–829 CrossRef CAS.
- F. Takeshita and T. Ochiya, Cancer Sci., 2006, 97, 689–696 CrossRef CAS.
- R. Quarck and P. Holvoet, Current Gene Therapy, 2004, 4, 207–223 CAS.
- T. C. Karagiannis and A. El-Osta, Cancer Gene Therapy, 2005, 12, 787–795 CrossRef.
- E. J. Sontheimer, Nat. Rev. Mol. Cell Biol., 2005, 6, 127–138 CrossRef CAS.
- D. Bumcrot, M. Manoharan, V. Koteliansky and D. W. Y. Sah, Nat. Chem. Biol., 2006, 2, 711–719 CrossRef CAS.
- S. Barik and V. Bitko, Expert Opinion on Biological Therapy, 2006, 6, 1151–1160 Search PubMed.
- S. Akhtar and I. Benter, Adv. Drug Delivery Rev., 2007, 59, 164–182 CrossRef CAS.
- M. A. Behlke, Molecular Therapy, 2006, 13, 644–670 Search PubMed.
- Y. Dorsett and T. Tuschl, Nat. Rev. Drug Discovery, 2004, 3, 318–329 CrossRef CAS.
- G. J. Hannon and J. J. Rossi, Nature, 2004, 431, 371–378 CrossRef CAS.
- A. Inoue, S. Y. Sawata and K. Taira, J. Drug Targeting, 2006, 14, 448–455 CrossRef CAS.
- C. X. Li, A. Parker, E. Menocal, S. L. Xiang, L. Borodyansky and J. H. Fruehauf, Cell Cycle, 2006, 5, 2103–2109 CAS.
- L. Aagaard and J. J. Rossi, Adv. Drug Delivery Rev., 2007, 59, 75–86 CrossRef.
- A. de Fougerolles, H. P. Vornlocher, J. Maraganore and J. Lieberman, Nat. Rev. Drug Discovery, 2007, 6, 443–453 CrossRef CAS.
- F. Y. Xie, M. C. Woodle and P. Y. Lu, Drug Discovery Today, 2006, 11, 67–73 CrossRef CAS.
- W. J. Kim, C. W. Chang, M. Lee and S. W. Kim, J. Controlled Release, 2007, 118, 357–363 CrossRef CAS.
- M. Golzio, L. Mazzolini, A. Ledoux, A. Paganin, M. Izard, L. Hellaudais, A. Bieth, M. J. Pillaire, C. Cazaux, J. S. Hoffmann, B. Couderc and J. Teissie, Gene Therapy, 2007, 14, 752–759 CrossRef CAS.
- S. Khanna, S. Roy, H. A. Park and C. K. Sen, J. Biol. Chem., 2007, 282, 23482–23490 CrossRef CAS.
- B. Goetze, B. Grunewald, S. Baldassa and M. Kiebler, J. Neurobiol., 2004, 60, 517–525 CrossRef CAS.
- T. A. Rand, S. Petersen, F. Du and X. Wang, Cell (Cambridge, MA, U.S.), 2005, 123, 621–629 Search PubMed.
- E. W. Song, P. C. Zhu, S. K. Lee, D. Chowdhury, S. Kussman, D. M. Dykxhoorn, Y. Feng, D. Palliser, D. B. Weiner, P. Shankar, W. A. Marasco and J. Lieberman, Nat. Biotechnol., 2005, 23, 709–717 CrossRef CAS.
- Y. Zhang, F. Calon, C. Zhu, R. J. Boado and W. M. Pardridge, Human Gene Therapy, 2003, 14, 1–12 Search PubMed.
- T. S. Zimmermann, A. C. H. Lee, A. Akinc, B. Bramlage, D. Bumcrot, M. N. Fedoruk, J. Harborth, J. A. Heyes, L. B. Jeffs, M. John, A. D. Judge, K. Lam, K. McClintock, L. V. Nechev, L. R. Palmer, T. Racie, I. Rohl, S. Seiffert, S. Shanmugam, V. Sood, J. Soutschek, I. Toudjarska, A. J. Wheat, E. Yaworski, W. Zedalis, V. Koteliansky, M. Manoharan, H. P. Vornlocher and I. MacLachlan, Nature, 2006, 441, 111–114 CrossRef CAS.
- K. Kataoka, K. Itaka, N. Nishiyama, Y. Yamasaki, M. Oishi and Y. Nagasaki, Nucleic Acids Symp. Ser., 2005, 49, 17–18 Search PubMed.
- X.-L. Wang, T. Nguyen, D. Gillespie, R. Jensen and Z.-R. Lu, Biomaterials, 2008, 29, 15–22 CrossRef CAS.
- B. R. Meade and S. F. Dowdy, Adv. Drug Delivery Rev., 2007, 59, 134–140 CrossRef CAS.
- P. Kumar, H. Q. Wu, J. L. McBride, K. E. Jung, M. H. Kim, B. L. Davidson, S. K. Lee, P. Shankar and N. Manjunath, Nature, 2007, 448, 39–U32 CrossRef CAS.
- S. Deshayes, M. C. Morris, G. Divita and F. Heitz, Cell. Mol. Life Sci., 2005, 62, 1839–1849 CrossRef CAS.
- L. Crombez, A. Charnet, M. C. Morris, G. Aldrian-Herrada, F. Heitz and G. Divita, Biochem. Soc. Trans., 2007, 35, 44–46 CrossRef CAS.
- J. Soutschek, A. Akinc, B. Bramlage, K. Charisse, R. Constien, M. Donoghue, S. Elbashir, A. Geick, P. Hadwiger, J. Harborth, M. John, V. Kesavan, G. Lavine, R. K. Pandey, T. Racie, K. G. Rajeev, I. Rohl, I. Toudjarska, G. Wang, S. Wuschko, D. Bumcrot, V. Koteliansky, S. Limmer, M. Manoharan and H. P. Vornlocher, Nature, 2004, 432, 173–178 CrossRef CAS.
- N. F. Bouxsein, C. S. McAllister, K. K. Ewert, C. E. Samuel and C. R. Safinya, Biochemistry, 2007, 46, 4785–4792 CrossRef CAS.
- S. L. Uprichard, FEBS Lett., 2005, 579, 5996–6007 CrossRef CAS.
- D. Kennedy, Science, 2002, 298, 2283–2283 CrossRef CAS.
- E. Goncalves, E. Kitas and J. Seelig, Biochemistry, 2005, 44, 2692–2702 CrossRef CAS.
- T. Jiang, E. S. Olson, Q. T. Nguyen, M. Roy, P. A. Jennings and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17867–17872 CrossRef CAS.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS.
- K. Huang, B. Voss, D. Kumar, H. E. Hamm and E. Harth, Bioconjugate Chem., 2007, 18, 403–409 CrossRef CAS.
- S. K. Hamilton, M. R. Ikizler, C. Wallen, P. F. Wright and E. Harth, Mol. BioSyst., 2008, 4, 1209–1211 RSC.
- S. K. Hamilton and E. Harth, ACS Nano, 2009, 3, 402–410 CrossRef CAS.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details for the assessment of fluorescence in confocal imaging. See DOI: 10.1039/c0py00342e |
| ‡ This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| § S.K.H. current address: School of Chemical and Bimolecular Engineering, Georgia Institute of Technology, 311 Ferst Dr NW, Atlanta GA, 30332, USA and Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, 313 Ferst Dr , Atlanta GA, 30332, USA |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.